




Ewolucja człowieka molekularnie Understand article
Tłumaczenie Jadwiga Schreiber. W swoim drugim artykule Jarek Bryk opisuje jak naukowcy analizują sekwencje DNA w celu zbadania molekularnych podstaw adaptacji

Henrik500/iStockphoto
DNA każdego organizmu zawiera informacje o jego ewolucyjnej historii. Badając DNA oraz zmiany w jego sekwencji – porównując sekwencje pomiędzy rożnymi osobnikami, populacjami lub pomiędzy gatunkami – możemy odkryć ich ewolucyjną przeszłość. Możemy dowiedzieć się, które geny czy fragmenty genomu mogły przyczynić się do adaptacji tych osobników, pozwalając im lepiej przetrwać i rozmnażać się (zobacz słowniczek dla wszystkich pogrubionych słów).
W poprzednim artykule (Bryk, 2010), opisałem kilka przykładów takich korzystnych zmian genetycznych u ludzi i innych organizmów. Udowodnienie, która zmiana genetyczna mogła być korzystna jest trudne – zwłaszcza u ludzi – ale jeszcze trudniejszym zadaniem jest znalezienie mechanizmu, dzięki któremu zmiany te mogły pozytywnie wpłynąć na przeżycie i reprodukcje organizmu.
W tym artykule prezentuję jeden ze sposobów, jaki naukowcy mogą użyć by, w pierwszej kolejności, móc zidentyfikować regiony naszego genomu, któreprawdopodobnie pomagały w przetrwaniu i reprodukcji, a następnie sprawdzić wjaki sposób regiony te mogły przynieść korzyści (adaptację) naszym przodkom.
Jednym ze sposobów, w jaki te potencjalnie korzystne fragmenty naszego genomu mogą zostać zidentyfikowane, jest po prostu porównanie sekwencji DNA wielu osobników z różnych populacji. W bardzo prostym przypadku, jeśli jedna z tych populacji znalazła się po wpływem presji selekcyjnej (na przykład silnego promieniowania UV), która nie wystąpiła u innych populacji, sekwencja DNA odpowiedzialna za tą adaptację (w tym wypadku ciemniejszy kolor skóry) powinna być różna.
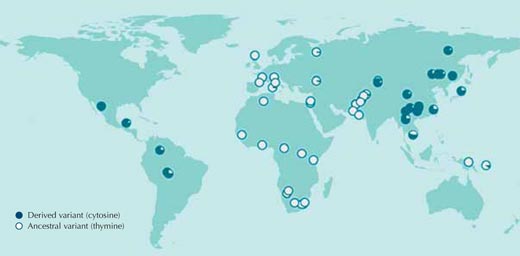
Rysunek zaadoptowany za pozwoleniem Macmillan Publishers Ltd: Nature, Sabeti et al. (2007), © 2007
W ogromnej większości przypadków jednak nie wiemy, jakiej presji selekcyjnej populacje zostały poddane w przeszłości, nie wiemy też, które sekwencje genetyczne są odpowiedzialne za adaptacje. Zacznijmy więc od porównania sekwencji DNA pomiędzy populacjami ludzi bez wstępnych założeń. Rysunek 1 przedstawia takie porównanie, przeprowadzone dla tylko jednego nukleotydu z genomu ludzkiego.
Jeśli osobniki posiadają różne nukleotydy w tej samej pozycji w sekwencji DNA, zjawisko to nazywamy polimorfizmem pojedynczego nukleotydu (single nucleotide polymorphism, w skrócie „SNP”, wypowiadane „snip”); trzy miliony takich wariantów w genomie ludzkim jest skatalogowanych w publicznie dostępnej bazie danych HapMapw1. SNP przedstawiony na rysunku 1, występuje w dwóch wariantach lub inaczej allelach: w tym miejscu w sekwencji jedną z dwóch zasad może być albo tymina (T) albo cytozyna (C).
Każdy okrąg na rysunku 1 reprezentuje pojedynczą populację i przedstawia częstość występowania jednego z dwóch możliwych allelów w danej populacji.
Allel zawierający tyminę obecny jest we wszystkich populacjach afrykańskich i większości europejskich ale nie występuje we wschodniej Azji i Amerykach, gdzie cytozyna jest bardziej rozpowszechniona (Sabeti et al., 2007, 2006; Xue et al., 2009).
Jeśli przeprowadzilibyśmy takie porównanie dla wszystkich trzech milionów SNP-ów z HapMap, zauważylibyśmy, że dystrybucja wariantów rs3827760 w populacjach ludzi jest bardzo nietypowa. Dlatego też rs3827760 zasługuje na szczególną uwagę, nawet jeśli jego rozmieszczenie nie mówi nam nic o potencjalnej korzyści jego wariantów (ich wartości adaptacyjnej), czy nawet o tym, czy są one w ogóle adaptacyjne. Jak na razie jedyne, co wiemy to to, że z jakiegoś powodu oryginalnie wystepująca w tym miejscu tymina, u naszych przodków w Afryce, zamieniona została na cytozynę i że zmiana ta rozprzestrzeniła się we wschodniej Azji i w Amerykach. Szacunki okresu, kiedy zmiana ta nastąpiła są bardzo niedokładne: wiemy, że gdzieś pomiędzy 1000 i 70000 lat temu wszystkie populacje we wschodniej Azji posiadały wariant cytozyny.
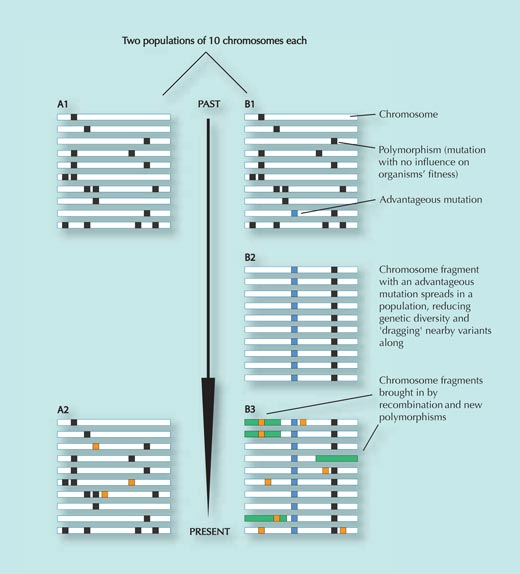
Populacja A nie doświadczyła pozytywnej selekcji i pozostała wyględnie niezmieniona wraz z upływem czasu, z wyjątkiem nabycia kilku przypadkowych zmian genetycznych, które nie wpłynęły na jej dostosowanie (pomarańczowe kwadraty w panelu A2; zmiany, które zmniejszają dostosowanie są usuwane z populacji) – porównaj panel A1 z panelem A2.
Populacja B natomiast przeniosła się w nowe środowisko, gdzie została ona skonfrontowana z nową presją selekcyjną. W tym nowym otoczeniu, zmiana genetyczna (niebieski kwadrat na panelu B1) przynosi korzyść osobnikom, ktore ją posiadają i szybko rozprzestrzenia się ona wśród populacji (gdyż osobiki z tą zmianą posiadają więcej potomstwa). Fragmenty DNA wsytępujące blisko tego SNP-u rozprzestrzeniają się razem z nim (im bliżej dwa różne nukleotydy są koło siebie, tym mniejsza szansa, że zostaną one rozdzielone podczas rekombinacji, kiedy fragmenty matczynego i ojcowskiego DNA się wymieniają – patrz rysunek 3).
Wynikiem szybkiego rozprzestrzenienia się konkretnej sekwencji DNA (SNP-u) wśród populacji jest zmniejszenie liczby różnych wariantów sekwencji w danym rejonie genomu, gdyż większość osobników będzie posiadało korzystny SNP razem z tymi samymi sekwencjami DNA występującymi koło tego SNP-u (porównaj panel B1 z panelem B2). Proces ten następuje stosunkowo szybko.
Po pewnym czasie jednak, nowe mutacje genetyczne oraz procesy rekombinacji wprowadzą nowe sekwencje DNA w tym regionie (zielone prostokąty i pomarańczowe kwadraty w panelu 3). Zatem, im więcej czasu upłynie od rozprzestrzenienia się korzystnej mutacji, tym trudniej jest ją zidentyfikować, gdyż wywołany przez nią spadek w różnorodności genetcznejw danym regionie genomu (B2) może zostać zamaskowany nowymi zmianami gentycznymi (B3)
Zdjęcie dzięki uprzejmości Jarek Bryk
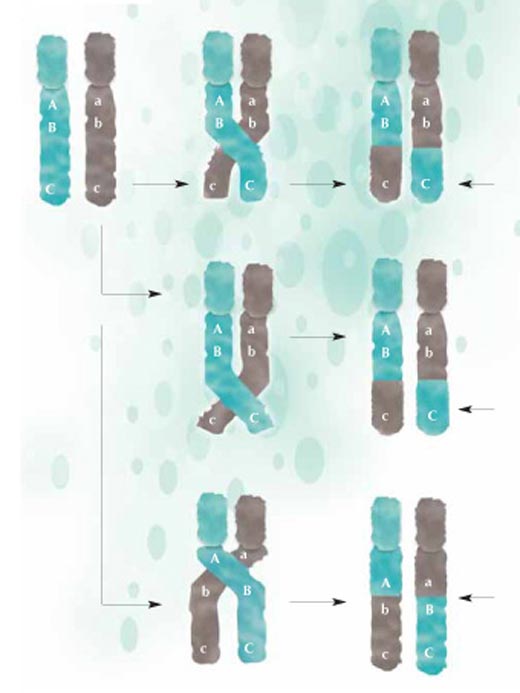
rekombinacji fragmenty DNA
wymieniane są pomiędzy
chromosomami matki
(zielone) i ojca (szare), a ich
nowe konfiguracje
przekazywane następnemu
pokoleniu. Im bliżej siebie
położone są różne sekwencje
gentyczne, tym mniej
prawdopodobne, że zostaną
one rozdzielone podczas
rekombinacji: A i B są bliżej
siebie niż B i C, zatem jest
mniej prawdopodobne że A i
B zostaną rozdzielone. Kliknij
na obrazek aby powiększyć
Zdjęcie dzięki uprzejmości
Nicola Graf
Jak zatem możemy zdecydować czy ta nowa wersja DNA powstała z wyniku pozytywnego doboru naturalnego (tzn. czy posiadanie cytozyny było korzystne we wschodniej Azji i w Amerykach) czy po prostu był to przypadek? Aby zbadać czy zmiana w DNA (tymina zamieniona na cytozynę) była wynikiem pozytywnego doboru naturalnego, przypatrzmy się sekwencjom DNAwokół SNP-u. Jeśli sekwencje DNA otaczające rs3827760 są podobne we wszystkich populacjach, mielibyśmy prawo przypuszczać, że ten SNP nie miał wpływu na dostosowanieorganizmu. Jeśli jednak jedna z populacji (np. wschodnio azjatycka) poddana została prescji selekcyjnej a rs3827760 przyczynił się do powstania adaptacji do tego czynnika selekcyjnego, sekwencje DNA otaczające SNP powinny różnić się między populacjami. Aby zrozumieć dlaczego tak jest, spójrzmy na rysunek 2.
Po porównaniu sekwencji wokół rs3827760 jest jasne, że różnorodność wokól wariantu cytozyny w populacjach wschodnio azjatyckich jest dużo mniejsza niż różnorodność wokół wariantu z tyminą, znalezionego w populacjach afrykańskich i europejskich (populacje amerykańskie nie były testowane). Sugeruje to, że pozytywny dobór spowodował rozprzestrzenienie się odmiany zawierającej cytozynę w populacjach wschodnio azjatyckich. Ale czy to naprawdę ten właśnie SNP został wybrany w drodze selekcji? – czy on w ogóle może być odpowiedzialny za zmianę w fenotypie organizmu?
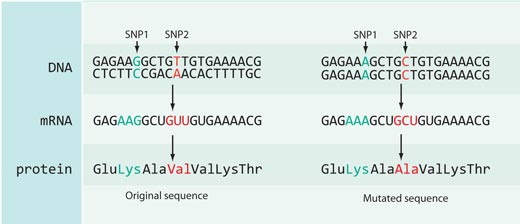
Nie wszystkie zmiany w sekwencji DNA mają wpływ na sekwencję białka: większość SNP-ów skatalogowanych w bazie danych HapMap występuje w niekodujących częściach genomu (np. między genami) albo są synonimiczne – tzn. znajdują się one w kodującym rejonie genomu ale nie powodują zmiany w sekwencji białka (zobacz rysunek 4).
Zdjęcie dzięki uprzejmości Jarek Bryk
W przypadku rs3827760 mieliśmy jednak szczęście, ponieważ SNP ten umiejscowiony jest w kodującej części genu – przy końcu genu zwanego edar, zaangażowanego w rozwój mieszków włosowych, gruczołów potowych i zębów. Co więcej, zmiana tyminy na cytozynę w sekwencji DNA powoduje zmianę sekwencji białka: Afrykanie i Europejczycy (posiadający wariant SNP-u z tyminą) zawierają w tym białku w pozycji 370 aminokwas walinę, podczas gdy populacje wschodnio azjatyckie i amerykańskie (z nukleotydem cytozynowym) mają w tej pozycji alaninę. Ta część białka EDAR odgrywa rolę w interakcjach z innymi białkami i jej mutacje powodują dysplazje ektodermalne – zaburzenia rozwojowe zębów, włosów i gruczołów potowych – u ludzi oraz myszy (zobacz rysunek 5). Fakt ten sugeruje, że zmiana aminokwasu w pozycji 370 może nie tylko zmienić sekwencję białka ale również jego funkcję, wpływając na funkcję organizmu.
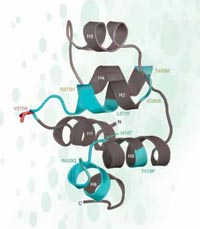
struktura fragmentu białka
EDAR. Mutacje zaznaczone na
zielono podowują u ludzi
dysplazję ektodermalną.
Położenie zmiany
powodowanej przez SNP
rs3827760 zaznaczony
zaznaczone jest na czerwono
Adapted by with permission
from Macmillan Publishers Ltd:
Nature, Sabeti et al. (2007), ©
2007
Aby sprawdzić czy zmiana w sekwencji białka rzeczywiście wpływa na jego funkcję musimy przejść do eksperymentów na poziome szlaków biochemicznych, w których białko EDAR bierze udział: serii reakcji chemicznych zaangażowanych w rozwój mieszków włosów, gruczołów potowych i zębów. Gdy szlaki te sprawdzono w laboratorium, w hodowlach komórkowych, wariant tego białka zawierający alaninę (występujący w populacjach we wschodniej Azji i Amerykach) bardziej aktywował szlak biochemiczny EDAR w porównaniu z wersją białka zawierającą walinę (występującą u populacji afrykańskiej i europejskiej). Wynik ten zgodny jest z danymi dotyczącymi sktruktury włosów, które wykazały, że ludzie z alaninowym wariantem białka EDAR mają grubsze włosy w porównaniu z ludzmi posiadającymi wariant walinowy. By jednak uzyskać bardziej bezpośredni dowód tej współzależności, myszy laboratoryjne zostały genetycznie zmodifikowane w celu zwiększenia poziomu aktywności zależnego od EDAR szlaku biochemicznego. Myszy te miały wyraźnie gęstsze futro i grubsze włosy, jak również m.in. większe gruczoły ślinowe w porównaniu z myszami z nie zmienionym poziomem aktywości tego szlaku (Chunyan et al., 2008; Chang et al., 2009).
Podsumowując, wyniki te sugerują, że dwa allele genu edar (tymina albo cytozyna) mogą wpływać na strukturę i funkcję białka EDAR oraz mogą prowadzić do wystąpienia różnic w wyglądzie i funkcji organizmów myszy i ludzi: różnic w grubości włosów oraz wielkości gruczołów wydzielniczych.
Różnice w sekwencjach DNA, które dziś obserwujemy są jednak jedynie historycznym zapisem eksperymentów, jakie zaszły w przyrodzie. Teraz możemy tylko spekulować na temat presji selekcyjnej, jakiej populacje azjatyckie i amerykańskie zostały poddane i która spowodowała rozprzestrzenienie się cytozynowego allelu. Połączenie badań genetycznych, doświadczeń laboratoryjnych i analizy fenotypów zwierząt modelowych i populacji ludzi i dzikich zwierząt pozwala nam przetestować hipotezy na temat funkcji różnic genetycznych występujących pomiędzy populacjami czy gatunkami. Dzięki takiemu podejściu, możemy starać się odkryć molekularne podstawy adaptacji oraz zrozumieć, w jaki sposób organizmy przystosowują się do ciągle zmieniającego się środowiska.
Słowniczek
Adaptacja: dana cecha jest adaptacją jeśli umożliwia ona osobnikowi lepiej przetrwać i reprodukować się w danym środowisku w porównaniu z osobnikami, które tej cechy nie posiadają. Bardziej formalnie, dana cecha jest adaptacją jeśli zwiększa dostosowanie (z ang. fitness) osobnika.
Dostosowanie (z ang. fitness): trudny do zdefiniowania termin z dziedziny biologii ewolucyjnej i genetyki populacji; określa on średnią liczbę potomstwa w ciągu jednego pokolenia przypadającą na konkretny genotyp w porównaniu z innym genotypem w populacji. Genotypy, które pozostawiają więcej potomstwa są lepiej dostosowane.
Genom: całkowite DNA danego organizmu. Zazwyczaj odnosi się to do jądrowego DNA u organizmów eukariotycznych, w przeciwieństwie doale obejmuje też DNA mitochondrialnego czy plastydowe, lub całość materiału genetycznego organizmów prokariotycznych i wirusówgo DNA. Więcej informacji na ten temat można znaleźć w „What is a genome” na stronie internetowej Amerykańskiej Nacjonalnej Biblioteki (US National Library)w2.
Dobór naturalny: jest jednym z mechanizmów ewolucji, opisuje ona różnice w przetrwaniu i reprodukcji osobników w danym środowisku. Dobór naturalny jest ‘pozytywny’, kiedy promuje cechę pozwalając osobnikom, które ją posiadają, lepiej przetrwać i rozmnażać się niż osobnikom nie posiadającym tej cechy.
Presja selekcyjna: czynnik środowiskowy (np. temperatura, występowanie pasożytów, drapieżnictwo lub agresja ze strony osobników tego samego gatunku) prowadzący do zróżnicowania szansy przeżycia i reprodukcji organizmów.
SNP: poliformizm pojedynczego nukleotydu, tznkiedy. pojedynczy nukleotydPojedyncza (litera w sekwencji DNA) różni się pomiędzy osobnikami lub chromosomami (. Wwymawia się “snip”).
References
- Bryk J (2010) Dobór naturalny molekularnie. Science in School 14: 58-62.
- Chang SH et al. (2009) Enhanced EDAR signalling has pleiotropic effects on craniofacial and cutaneous glands. PLoS ONE 4(10): e7591. doi: 10.1371/journal.pone.0007591
- W artykule tym opisano fenotypy różnych gruczołów myszy ze zwiększoną aktywnością białka EDAR oraz spekulujacje na temat cech, które ukształtowane zostały pod wpływem doboru naturalnego w historii człowieka. Artykuł ten dostępny jest bezpłatnie na stronie internetowej magazynu: www.plosone.org
- Chunyan M et al. (2008) Enhanced ectodysplasin-A receptor (EDAR) signaling alters multiple fiber characteristics to produce the East Asian hair form. Human Mutation 29(12): 1405-1411. doi: 10.1002/humu.20795
- Artykuł ten szczegółowo opisuje i ilustruje badania in vitro białka EDAR oraz transgenicznych myszy.
- Pongsophon P, Roadrangka V, Campbell A (2007) Counting Buttons: demonstrating the Hardy-Weinberg principle. Science in School 6: 30-35.
- Sabeti PC et al. (2006) Positive natural selection in the human lineage. Science 312(5780): 1614-20. doi: 10.1126/science.1124309
- Jest to znakomity przegląd różnych metod używanych do badań doboru naturalnego selekcji na poziomie genomu.
- Sabeti PC et al. (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449: 913-918. doi: 10.1038/nature06250
- Można bezpłatnie pobrać ten artykuł tutaj, lub zasubskrybować Nature: www.nature.com/subscribe
- Artykuł ten opisuje jeden ze sposobów analizy genomu w poszukiwaniu śladów doboru naturalnego.
- Xue Y et al (2009) Population differentiation as an indicator of recent positive selection in humans: an empirical evaluation. Genetics 183(3): 1065-77. doi: 10.1534/genetics.109.107722
- Artykuł ten zawiera dyskusję o edar i innych podobnych genach. Dostępny jest on bezpłatnie przez stronę PubMed Central: www.ncbi.nlm.nih.gov/pmc lub bezpośrednio przez link: http://tinyurl.com/26xte2h
Web References
- w1 – Projekt HapMap to zrzeszenie naukowców i organizacji finansujących te badania z Kanady, Chin, Japoni, Nigerii, Wielkiej Brytani oraz USA w celu zorganizowania publicznie dostępnej bazy danych, która ułatwi naukowcom znaleźć geny związane z chorobami u ludzi oraz badać podłoże różnych reakcji na leki: www.hapmap.org
- w2 – Więcej informacji na temat genomu i Projektu ‘Human Genome Project’ jest dostępnych w ‘What is a genome’ na stronie internetowej US National Library of Medicine: http://ghr.nlm.nih.gov/handbook/hgp/genome
Review
Mimo całej naszej wiedzy o sekwencji ludzkiego genomu, precyzyjne funkcje wielu jego fragmentów oraz jak i dlaczego sekwencje DNA zmieniały się w populacjach są wciąż w dużej mierze niewyjaśnione. Adaptacje ewolucyjne u ludzi wystąpiły na pewno, ale trudno jest je udowodnić. Artykuł ten opisuje jak takie zmiany są identyfikowane. Doświadczenia z genetycznie zmodifikowanymi myszami demonstrują jak zmiana pojedyńczego nukleotydu w DNA, która zmienia sekwencję aminokwasową w białku, prowadzi do zmiany w strukturze i funkcji tego białka. To z kolei może spowodować zróźnicowanie morfologiczne (fenotypowe) osobników.
Na lekcjach biologii, artykuł ten mógłby być wykorzystany do omówienia takich tematów jak: częstotliwości używania poszczególnych kodonów oraz kodonów zdegenerowanych; sutruktury i funkcji białek oraz genetyki populacyjnej. Może on również służyć do zdobycia podstawowej wiedzy na temat zróżnicowania populacji ludzkich oraz jako wprowadzenie do opisanych badań w Sanger Institute oraz Human Genome Mapping Project.
Uczniowie mogą przeprowadzić dyskusję na temat adaptacji ewolucyjnych, referując konkretne przykłady podane w tym artykule. To mogłoby również skierować dyskusję na temat selekcji naturalnej, genetyki populacyjnej oraz równowagi Hardiego-Weinberga. Dyskusja ta mogłaby być uzupełniona w oparciu o wspaniały artykułu w wydaniu 6 Science in School (Pongsophon et al., 2007).
Odpowiednimi pytaniami sprawdzającymi zrozumienie tamatu mogłyby być:
- Opisz we własych słowach co to jest i podaj przykład (inny niż podany w tekście artykułu) SNP-u.
- Wyjaśnij znaczenie SNP-ów.
- Który aminokwas kodowany jest przez nukelotyd GTT?
- Opisz zmiany zaobserwowane u mysz z genetycznie zmodyfikowym szlakiem biochemicznym zależnym od EDAR i zaproponuj w jaki sposób zaobserwowane zmiany mogą zostać skwantyfikowane.
Shelley Goodman, Wielka Brytania