Supporting materials
Tables for students (Word document)
Download
Download this article as a PDF





Tradotto da Monica Menesini. La Bioinformatica in generale richiede potenti computer; con l’aiuto di Cleopatra Kozlowski potrete invece investigare il nostro antenato primate armati soltanto di carta e penna.
In seguito agli ultimi sviluppi tecnologici, è relativamente semplice e veloce determinare una sequenza di DNA o di una proteina. Queste sequenze tuttavia di per sé ci dicono ben poco: GAATCCA, per esempio. Abbiamo bisogno di conoscere il significato delle sequenza. Quali proteine sono codificate da quella sequenza di DNA? O la sequenza, invece, non codifica per una proteina? Che effetti avrà un piccolo cambiamento nel DNA sulla struttura della proteina? Che funzione svolge quell proteina nella cellula? E, naturalmente, cosa ci può dire quella certa sequenza di DNA sulla nostra storia evolutiva?
Queste ed altre importanti questioni possono essere risolte con la bioinformatica, essenzialmente paragonando le sequenza di DNA o di proteine – per esempio paragonando una sequenza scoperta da poco con sequenze per le quali abbiamo già molte informazioni (forse hanno la stessa funzione?) o paragonando sequenze simili in specie diverse.
La bioinformatica generalmente richiede l’uso di potenti computer. E’ troppo semplice però lasciar fare al computer tutto il lavoro senza comprendere cosa c’è sotto. Per questo motivo, queste attività sono progettate per essere svolte su carta, in modo che gli studenti capiscano come funziona l’analisi bioinformatica.
Questo articolo riguarda una di un gruppo di quattro attività. Le due attività introduttive (“trovare un gene” e “mutazioni”) e quella conclusiva (“DNA mobile”) possono essere scaricate dal sito dell’European Learning Laboratory for the Life Sciences (ELLS)w1. Tutte le tabelle necessarie agli studenti per completare questa attività, la procedura in dettaglio e le risposte alle domande di comprensione possono essere scaricate dal sito di Science in Schoolw2.
L’accumulo di mutazioni produce cambiamenti del DNA nel corso delle generazioni. Questa attività dimostra come questo possa essere utilizzato per dedurre le relazioni evolutive tra gli organismi. Richiede circa 90 minuti e fa uso solo di penna e tabelle scaricabili dal sito di Science in Schoolw2.
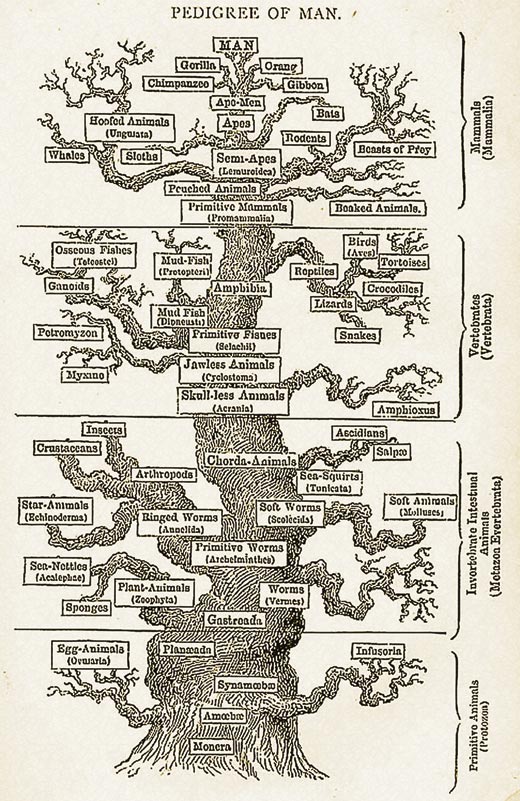
Pensate a come classifichereste diversi animali. Tradizionalmente sono state usate le differenze fisiche per determinare le relazioni evolutive tra di loro, per esempio se l’organismo ha una colonna vertebrale o se ha le ali. Questo approccio però può causare dei problemi. Per esempio gli uccelli, i pipistelli e gli insetti hanno le ali, ma sono strettamente imparentati? Come si misura il tempo trascorso dalla divergenza rispetto all’antenato comune?
Sappiamo dagli studi di sequenziamento del DNA che le mutazioni avvengono casualmente ad un ritmo molto lento e vengono trasmesse dai genitori alla prole. Così, se assumiamo che tutti gli organismi hanno un antenato comune, si possono usare le differenze nelle sequenze omologhe per misurare quanto tempo è passato da quando gli organismi si sono separati. In altre parole, più lontano nel tempo è il momento della divergenze, più le sequenze di DNA saranno diverse.
Le sequenze omologhe sono quelle sequenze che hanno un’origine comune. In realtà non possiamo sapere con certezza se due sequenze sono omologhe (non eravamo lì a vedere il DNA cambiare nel tempo) ma se sono sufficientemente simili possiamo assumere che siano omologhe. Per determinare quanto simili sono due sequenze, vanno allineate correttamente (ma questo non fa parte di questa attività).
Da notare che regioni diverse del DNA – codificanti e non codificanti – evolvono a velocità diverse. In generale, le regioni codificanti evolvono più lentamente, perché una mutazione che produce un cambiamento in una proteina è generalmente più dannosa per l’organismo che quindi ha meno probabilità di sopravvivere e riprodursi. Questo è discusso nell’attivita “DNA mobile”.
Per illustrare il concetto di omologia si può usare l’esempio della filologia – lo studio dell’evoluzione delle lingue. Ci sono infatti molti parallelismi tra i metodi usati per studiare l’evoluzione delle lingue e degli organismi.
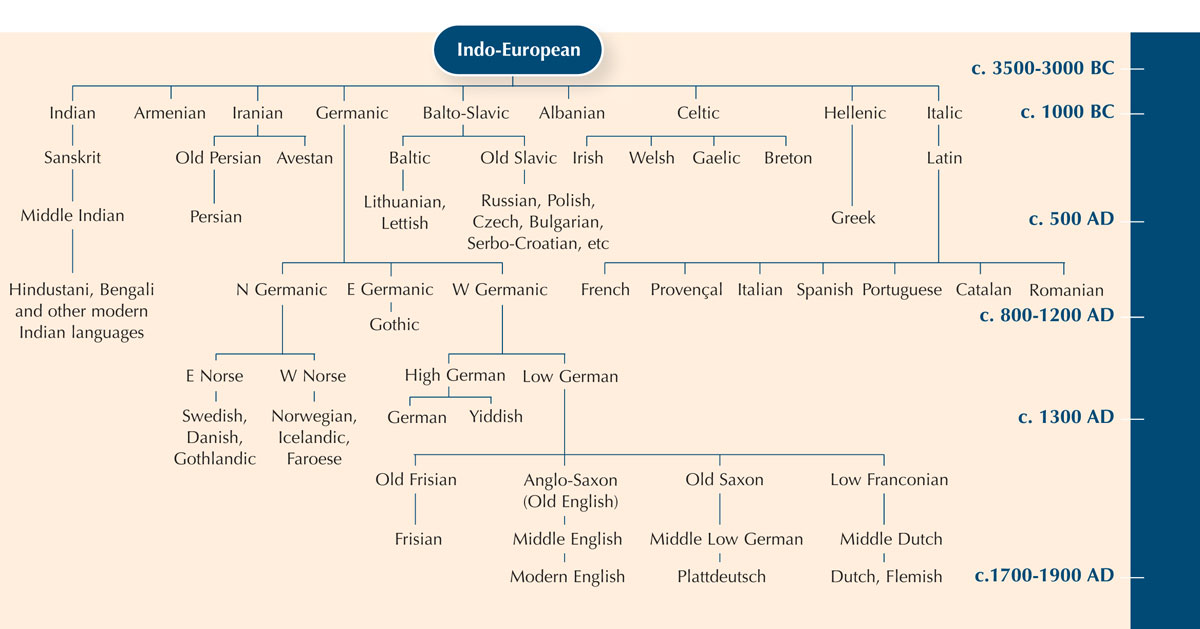
Usare le differenze tra frammenti di sequenze di DNA è un po’ come paragonare una parola che ha lo stesso significato nelle varie lingue, per scoprire quanto strettamente sono imparentate.
| Armeno | gatz |
| Basco | katu |
| Olandese | kat |
| Inglese | cat |
| Estone | kass |
| Finlandese | kissa |
| Islandese | kottur |
| Italiano | gatto |
| Norvegese | katt |
| Polacco | kot |
| Portoghese | gato |
| Russo | kot |
| Spagnolo | gato |
| Svedese | katt |
Si può notare che la parola “gatto” in Italiano, Spagnolo e Portoghese è più o meno la stessa: gatto, gato e gato. Sia in Svedese che in Norvegese la parola è “katt”, ma in Finlandese è diversa: “kissa”. Sebbene la Finlandia, così come la Svezia e la Norvegia, sia un paese Nordico, il termine Finlandese per “gatto” è più simile al termine Estone, “kass. Infatti le due lingue sono strettamente legate. Si può quindi imparare qualcosa sulla relazione tra le lingue studiando come le parole sono cambiate nel tempo.

In questa attività costruiremo un albero filogenetico utilizzando 5 sequenze omologhe di DNA di primati. Poiché le sequenze sono fittizie, non possiamo ricavare una stima sicura della distanza genetica; per farlo avremmo bisogno di usare sequenze molto più lunghe. Nonostante ciò, le sequenze finte (in tabella 2) sono state scelte per fornire un quadro sufficientemente accurato delle relazioni tra primati.
Nota: tutte le tabelle necessarie per il completamento di questa attività da parte degli studenti possono essere scaricati dal sito Science in Schoolw2.
| Primate | Sequenza |
|---|---|
| Neanderthal (n) | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
| Uomo (h) | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Scimpanzé (c) | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
| Gorilla (g) | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
| Orangutan (o) | ACAACCTGCACTCCTATTCTGCCGAGCCGGGCGCGTGGCAAAGTCC |
| Neanderthal | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
|---|---|
| Uomo | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Scimpanzé | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
|---|---|
| Gorilla | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
Le tabelle di confronto di tutte le coppie di specie e la tabella completa delle differenze (Tabella 4) possono essere scaricate dal sito Science in Schoolw2.
| Neanderthal | Uomo | Scimpanzé | Gorilla | Orangutan | |
|---|---|---|---|---|---|
| Neanderthal | 0 | 3 | |||
| Uomo | 3 | 0 | |||
| Scimpanzé | 0 | 11 | |||
| Gorilla | 11 | 0 | |||
| Orangutan |
Il numero delle differenze fra due sequenze diviso per il numero totale di nucleotidi in ogni sequenza (in questo caso 46) ci fornisce il dato relativo alla distanza proporzionale tra le due sequenze
La “sequenza media”tra due specie viene presa come sequenza dell’antenato comune. In questo esercizio, non calcoliamo direttamente la sequenza media di, per esempio, Neandetthal e Uomo, ma la distanza evolutiva tra l’antenato comune di Neanderthal e Uomo e tutti gli altri primati del gruppo.
| Neanderthal/ Uomo | Scimpanzé | Gorilla | Orangutan | |
|---|---|---|---|---|
| Neanderthal/ Uomo | 0 | (4+5)/2 = 4.5 | (11+12)/2=11.5 | |
| Scimpanzé | (4+5)/2 = 4.5 | 0 | ||
| Gorilla | (11+12)/2=11.5 | 0 | ||
| Orangutan |
Usando la Tabella 5, cominciare a costruire l’albero evolutivo
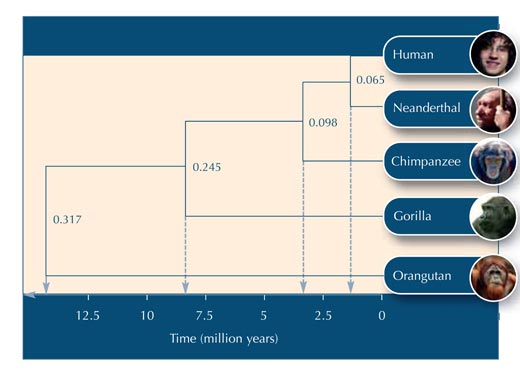
Di seguito alcune domande da usare per valutare la comprensione dell’attività da parte degli studenti. Le risposte possono essere scaricate dal sito Science in Schoolw2.
Questa attività è stata sviluppata in collaborazione tra l’ European Learning Laboratory for the Life Sciences (ELLS)w1 e l’ European Molecular Biology Laboratory’s E-STAR Fellows per produrre risorse didattiche per le scuole. Cleopatra Kozlowski ha usufruito di una borsa di studio E-STAR finanziata da European Commission’s Framework Programme 6 Marie Curie Host Fellowship for Early Stage Research Training, contratto n. MEST-CT-2004-504640.
Hodge R (2006) A new tree of life. Science in School 2: 17-19.
Quando pensiamo alla bioinformatica probabilmente immaginiamo potenti computer e macchine sequenziatrici, ma i metodi di questa nuova scienza possono essere presentati in classe attraverso semplici attività da svolgere con carta e penna, come fa Cleopatra Kozlowski in questo articolo.
L’autrice ci sfida con la costruzione dell’albero filogenetico degli umani e altri primati sulla base delle differenze genetiche fra brevi sequenze (false) di DNA. L’attività proposta può essere sfruttata in modo divertente nelle scuole secondarie per affrontare alcune tematiche biologiche complesse come gli orologi molecolari nello studio dell’evoluzione.
L’articolo è rivolto agli insegnanti di scienze, che troveranno utili esercizi di comprensione alla fine del testo; gli studenti potranno utilizzare le domande per approfondire la loro comprensione dell’argomento. Le fonti sul web forniscono ulteriori informazioni e risorse.
Giulia Realdon, Italia
Tables for students (Word document)
Download this article as a PDF