




Deadly proteins: prions Understand article
Since the epidemic of ‘mad cow disease’ in the 1980s and 90s, and the emergence of its human equivalent, variant Creutzfeld-Jacob disease, there has been a great deal of research into prions, the causative agents. Mico Tatalovic reviews the current state of knowledge.

/ iStockphoto
There may be a hidden epidemic waiting to occur, with millions of people already infected; we cannot prevent or cure it, and we cannot even diagnose it until the fatal symptoms appear.
The disease is variant Creutzfeld-Jacob disease (vCJD), one of a group of diseases known as transmissible spongiform encephalopathies (TSEs), which are caused and transmitted by abnormal forms of prion proteins. Examples of TSEs include not only vCJD, but also scrapie in sheep, bovine spongiform encephalopathy (BSE or mad cow disease) in cattle and kuruw1 in humans. These diseases create large fluid-filled holes in the brain tissue because the accumulation of aberrant prions causes the neurons (nerve cells) to die. It is these characteristic holes that give the diseases their names: spongiform (sponge-like) encephalopathies (brain diseases).
TSEs affect the central nervous system, with symptoms including problems with co-ordination and balance, shakiness and uncontrollable jerking movements. In humans, TSEs also cause personality changes and depression, and sufferers may experience confusion, memory problems and insomnia. As the diseases progress, most mental functions are lost, including the ability to speak. All TSEs are fatal, and as yet, there is no cure.

proteins (orange) from an animal
infected with scrapie
Image courtesy of R. Dourmashkin
/ Wellcome Images
Prions are specific proteins found mainly in the nervous system, where – in their normal forms – they may have important functions. For example, studies on sea slugs, Aplysia, suggest that prions have a crucial role in memory formation (Si et al., 2010). Infectious prions are abnormal (aberrant) forms of prion proteins that replicate inside the host by forcing normal proteins of the same type to adopt the aberrant structure. This has a domino effect whereby a small number of aberrant prions can affect many normal ones and eventually lead to disease. As the aberrant prions form amyloids – aggregates of protein – in the cells, the cells die, creating holes in the brain.
Prions are the only known case of self-propagating pathogenic (disease-causing) proteins, and they are able to cause severe illness even though they seem to be just protein molecules: unlike bacteria, viruses or other known pathogens, they have no information encoded in nucleic acids (DNA or RNA) about how to invade and replicate within the host. There is still a veil of mystery around prions and exactly how they replicate, cross the blood-brain barrier and cross the species barrier – i.e. infect different species of host.
It was in the 1960s that investigators first found that TSE disease-causing agents appeared to lack nucleic acids; Tikvah Alper suggested that the agent was a protein. This idea sounded heretical because all other known disease-causing agents contained nucleic acids and their virulence and pathogenesis were genetically determined.

Robert Koch (1843-1910)
However, three decades of subsequent investigations, pursued most notably by Stanley Prusiner, who was awarded the Nobel Prize in Physiology or Medicine in 1997 for his work with prions and TSEs, resulted in the wide acceptance of this ‘protein-only hypothesis’w2.
Nevertheless, there are still those who believe that prion diseases are actually caused by unconventional viruses and that prion proteins are just part of this mysterious virus. Koch’s postulates describe what is needed to prove that a certain agent causes a particular disease; one of the necessary steps is to use that agent to induce the disease in a healthy organism. To prove that a TSE is indeed caused by the prion protein itself, the isolated, purified aberrant prions must be used to transmit the disease. It wasn’t until February 2010 that exactly this was done, adding further substantial evidence for the protein-only hypothesis (Wang et al., 2010).
Why do some scientists fear a vCJD epidemic?
The most worrying prion strain is the one that causes vCJD – a form of mad cow disease that has crossed the species barrier to infect humans (see box below). The first case of vCJD appeared in the UK in 1996 and has since killed 168 people there, with a peak in 2000 when 28 people diedw3, and there have been small numbers of cases in other countries including France, Italy, Ireland, Canada and the USA. Exact numbers of people infected or dying from this disease around the world are not known and although there is still uncertainty about how people become infected, the main infection route is thought to be through eating infected beef.
| UK | 172 (4) |
|---|---|
| France | 25 (0) |
| Republic of Ireland | 4 (0) |
| Italy | 2 (1) |
| USA | 3 (0) |
| Canada | 1 (0) |
| Saudi Arabia | 1 (1) |
| Japan | 1 (0) |
| Netherlands | 3 (0) |
| Portugal | 2 (0) |
| Spain | 5 (0) |
Most other prion diseases, including ‘normal’ CJD, are genetic or occur for unknown reasons, and affect elderly people. Variant CJD differs from CJD in that it affects young people and the aberrant prions are found not only in the brain but also in tissues such as blood, tonsils and appendices; this opens new ways of transmission to the entire population and not only the elderly.
For example, prions could be spread through blood transfusions. In the UK, donated blood is leucodepleted (the leucocytes, which may harbour infectious prions, are removed). Many other countries (e.g. Germany) ban blood donations from people who have lived in the UK.
The disease may also be spread by surgical instruments: infectious prions are resistant to high temperatures, irradiation and common chemical treatments that destroy other known pathogens. Estimates of how serious the risk is vary.
Furthermore, prions have also been found in the epididymis and seminal plasma tissues of rams (Gatti et al., 2002). Such findings led to concern about prion infection via sperm donations and even resulted in a US ban on sperm donations by Europeans and men who have lived in Europe. Nonetheless, a survey of experts worldwide estimated that the chance of prion transmission via sperm was less than 1:10 000 000 (Mortimer & Barratt, 2006).
But tissue contact – whether via sperm or blood or on surgical instruments – is not the only potential source of transmission: most of us consume milk and dairy products.
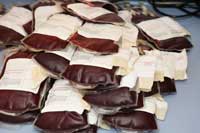
Image courtesy of choja / iStockphoto
Image courtesy of UCL /
Wellcome Images
Image courtesy of ImageMediaGroup / iStockphoto

infection: milk
Image courtesy of DaveAlan
/ iStockphoto
In 2006, a team of scientists from Switzerland detected low levels of the normal form of prions in milk bought in European shops (Franscini et al., 2006). Another study found that aberrant prions were able to replicate within mammary glands of scrapie-infected sheep (Ligios et al., 2005). Taken together, these results suggest that aberrant prions might be present in the milk of animals suffering from prion diseases. As prion disease symptoms take several years to develop, aberrant prions may be present in milk that is sold before the animals are diagnosed with the illness. Nonetheless, although it is not yet possible to give a clear estimate of the risk, it is generally thought that milk is safe until proven otherwise.
It seems, therefore, that we could contract vCJD via several routes although the risk is probably low. But are we all equally at risk? Several studies have shown that vCJD only affects people with certain variants (alleles) of a particular gene (prnp); other people seem to be resistant or do not develop the symptoms as fastw4. Worryingly, about 40% of the Western European and North American population and up to 92% of the Japanese population are homozygous for these susceptibility alleles. Furthermore, research on kuru suggests that even individuals without the susceptibility allele may become infected but simply take much longer to develop symptoms. So perhaps we are all susceptible to prion infection.
Because of all of these possible transmission routes and uncertainties in how important they are, the results of studies predicting the number of people dying from vCJD in the UK range from several hundred to more than ten million people. Even though it now looks as though the vCJD epidemic in the UK is diminishing, this may be just because many infected people are still developing symptoms. No one knows for sure. Ongoing research will elucidate just how risky prion diseases are to public health.
BSE (mad cow disease)
BSE came to scientists’ attention in 1986 when the first cases of a new neurological disease in cattle were discovered in the UK. The cause was traced to the use of beef products in cattle feed. In the resulting BSE epidemic, 181 376 cases of BSE were recorded in the UK by November 2002 and millions of cattle were culled to halt the epidemic. The first cases outside the UK appeared in 1989 and since then there have been several thousand cases in other countries, including most of the European countries, the USA, Canada, Japan and Israel. However, cattle feed with bovine parts was banned in the UK in 1988 and there are strict monitoring procedures in place to keep track of BSE.
Cases of BSE peaked in the UK in 1992 (37 280 cases) and have decreased dramatically since then (12 cases in 2009). The number of cases in other countries peaked some years later (2001-03) but has also decreased dramatically since thenw5.
References
- Franscini N et al. (2006) Prion protein in milk. PLoS ONE 1: e71. doi: 10.1371/journal.pone.0000071
- All PLos ONE articles are freely available online.
- Gatti JL et al. (2002) Prion protein is secreted in soluble forms in the epididymal fluid and proteolytically processed and transported in seminal plasma. Biology of Reproduction 67: 393-400. doi: 10.1095/biolreprod67.2.393
- Ligios C et al. (2005) PrPSc in mammary glands of sheep affected by scrapie and mastitis Nature Medicine 11: 1137–1138. doi: 10.1038/nm1105-1137
- Mortimer D, Barratt CLR (2006) Is there a real risk of transmitting variant Creutzfeldt–Jakob disease by donor sperm insemination? Reproductive BioMedicine Online 13: 778–790
- To view articles in Reproductive BioMedicine Online, it is necessary to register, but registration is free. See: http://www.rbmojournal.com/
- Si K et al. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140: 421-435. doi: 10.1016/j.cell.2010.01.008
- Wang F et al. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327: 1132-1135. doi: 10.1126/science.1183748
Web References
- w1 – Information about kuru is available on the website of the US National Institute of Neurological Disorders and Stroke: www.ninds.nih.gov/disorders/kuru
- In 1976, Carleton Gajdusek received the Nobel Prize in Physiology or Medicine for his work on kuru. More information is available in the press release, his autobiography, Nobel Prize lecture and other materials on the Nobel Prize website. See: http://nobelprize.org/nobel_prizes/medicine/laureates/1976
- w2 – To learn more about Stanley Prusiner’s work, see the press release, his autobiography and other materials on the Nobel Prize website: http://nobelprize.org/nobel_prizes/medicine/laureates/1997
- w3 – Statistics on the reported cases of BSE and deaths from vCJD are available on the website of the European Centre for Disease Prevention and Control: (www.ecdc.europa.eu) or via the direct link: http://tinyurl.com/yjbx8tn
- w4 – To learn more about the genetic disposition to develop prion diseases, see the website of the prion unit at the UK’s Medical Research Council (www.prion.ucl.ac.uk) or use the direct link: http://tinyurl.com/yaqau4a
- w5 – Statistics on BSE and many other animal diseases are available on the website of the World Organisation for Animal Health: www.oie.int
Resources
- More information about prions is available on the websites of the US Centers for Disease Control and Prevention: www.cdc.gov/ncidod/dvrd/prions
- To learn more about BSE, see the website of the World Health Organization: www.who.int/zoonoses/diseases/bse/en
- Research papers that may be of interest include: Aguzzi A, O’Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery 9: 237-248. doi: 10.1038/nrd3050
- Collinge, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318: 930-936. doi: 10.1126/science.1138718
- Di Guardo G, Marruchella G (2010) Prions and neuronal death. Cell Death and Disease 1: e6. doi: 10.1038/cddis.2009.9
- Welberg L (2010) Prions: a protective role for prions. Nature Reviews Neuroscience 11: 151. doi: 10.1038/nrn2812
- Koch’s postulates were published in the late 19th century by Robert Koch. For a more recent discussion of the postulates and their validity, see:
- Evans AS (1976) Causation and disease: the Henle-Koch postulates revisited. The Yale Journal of Biology and Medicine 49: 175-195. The article can be downloaded free of charge from PubMed Central (www.ncbi.nlm.nih.gov/pmc) or via the direct link: http://tinyurl.com/y9t2pd8
Review
Many people have heard of vCJD or mad cow disease but know little about the causative agent and the disease itself. This article explains the disease and cites up-to-date research on the causative agent, a prion. This will be useful for the infectious diseases topic in many science curricula and for general knowledge in other subjects. The links to data on BSE could be used in mathematics lessons or for learning to draw relevant graphs from data. The article lends itself to discussions about food safety, the effect of the disease outbreak on the British beef farmers, as well as comprehension and extension work. Some comprehension ideas include:
- Over how many years have prions been researched?
- 10 years
- 25 years
- 30 years
- 40 years
- Who won a Nobel Prize for research into prions?
- What is a prion?
- Describe the effects of spongiform encephalopathies.
- Which tissues have prions been discovered in?
- How could prions be spread?
Ideas for extension work are:
- Research the BSE outbreak in the UK in the late 1980s and describe the measures that were introduced to limit it.
- State Koch’s postulates. Explain why prions did not fit into this hypothesis until recently.
Shelley Goodman, UK