




Wie man biologische Datenbanken verwenden kann um Evolution und Biochemie zu lehren Teach article
Übersetzt von Marie-Luise Winz. Online-Werkzeuge können genutzt werden, um Sequenzen von Proteinen zu vergleichen und um zu verstehen, wie verschiedene Organismen entstanden sind.
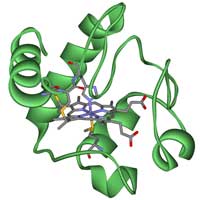
Cytochrom C
Mit freundlicher Genehmigung
von Klaus Hoffmeier/Wikimedia
commons
In der Vergangenheit führten Wissenschaftler evolutionäre Analysen durch, indem sie physische Eigenschaften verschiedener Spezies – auch Phänotypen genannt – in Fossilien verglichen. Seit der Entdeckung der molekularen Uhr hat sich dies alles jedoch verändert.
Das Konzept der molekularen Uhr ist aus der Beobachtung heraus entstanden, dass DNA und Proteine zweier Spezies umso unterschiedlicher sind, je länger ihre Abspaltung von einem gemeinsamen Vorfahren zurückliegt (für einen Übersichtsartikel, siehe Bromham & Penny, 2003).
Indem man homologe Gen- oder Proteinsequenzen vergleicht – in anderen Worten, die Sequenzen aus zwei Organismen mit einem gemeinsamen Vorfahren – kann man messen, wie viel Zeit vergangen ist, seitdem diese Organismen sich voneinander abgespalten haben. Dies kann in einem phylogenetischen Baum visualisiert werden.
Um zu untersuchen, wie ähnlich zwei Gene sind, muss man ihre Sequenzen kennen und diese korrekt alinieren (Kozlowski, 2010). Diese Sequenzen zu bekommen war früher sehr schwierig, heutzutage jedoch nicht mehr.
Ihre Schüler haben Ihnen sicher gesagt, dass alles im Internet steht – in diesem Falle haben sie Recht. Es gibt im Internet viele verschiedene frei zugängliche biologische Datenbanken, die echte Forschungsdaten enthalten. Für diese Übung werden wir jedoch zwei bestimmte Ressourcen verwenden.
Das Nationale Zentrum für Biotechnologie Information (National Center for Biotechnology Information – NCBI)w1 in Bethesda, MD, USA, bietet Zugang zu biomedizinischen und genomischen Informationen, während das Europäische Bioinformatik Institut (European Bioinformatics Institute – EBI)w2, das sich in Hinxton, UK, befindet, freien Zugang zu Daten aus lebenswissenschaftlichen Experimenten zur Verfügung stellt und Grundlagenforschung in ‘Computational Biology’, der computergestützten Biologie durchführt. Die NCBI Datenbank stellt Ihnen die Sequenz von jedem bisher sequenzierten Gen oder Protein zur Verfügung. Dann kann man die Werkzeuge vom EBI benutzen, um die Sequenzen zu alinieren und zu analysieren.
Übung
Wenn man die evolutionäre Verwandtschaft zwischen verschiedenen Organismen untersucht, ist es wichtig, die Gene oder Proteine, die dazu benutzt werden sollen, mit Bedacht auszusuchen. Es gibt einige wohlbekannte homologe Gene, die verwendet werden können, z.B. die für die Proteine Hämoglobin oder Cytochrom C, und in dieser Übung werden wir letzteres verwenden. Cytochrom C ist ein kleines Häm-Protein, das ein zentraler Bestandteil der Elektronentransportkette in den Mitochondrien ist. Alle aeroben Organismen stammen von einem gemeinsamen Vorfahren ab, der als erster Cytochrom C benutzt hat. Daher ist das Gen eine gute Wahl für unsere Zweckew3.
Diese Übung ist in drei Teile unterteilt:
- die Aminosäuresequenz von Cytochrom C in unterschiedlichen Organismen finden,
- diese alinieren, und
- einen phylogentischen Baum erstellen.
Am Ende sind noch einige Fragen angefügt, die bei der Untersuchung der evolutionären Verwandtschaften als Leitfaden dienen sollen.

klicken.
Proteinsequenzen finden
- Auf die NCBI Homepagew1 gehen.
- In der Suchmaske oben auf der Seite im Drop-down Menü ‘Protein’ auswählen.
- Den Namen der Spezies, z.B. Homo sapiens, und (auf Englisch) ‘cytochrome c’ eingeben.
- Den ‘Search’ Knopf klicken.
- Eine neue Seite wird nun die Resultate auflisten. Die meisten beinhalten dieselbe Sequenz aus unterschiedlichen Quellen, aber andere können auch partielle Sequenzen enthalten, oder zu einer anderen Spezies oder einem anderen Protein gehören. Mit Sorgfalt das richtige Protein aussuchen und auf den untenstehenden Link, der mit ‘FASTA’ beschriftet ist‚ klicken.
- Aus der Seite, die nun geladen wird, die Folge von Großbuchstaben herauskopieren, die die Aminosäuresequenz darstellt. Diese Buchstaben in ein Word Dokument kopieren, und dabei daran denken, die Namen von Sequenz und Organismus zu notieren.
- Dasselbe für so viele Organismen wie gewünscht durchführen, abhängig davon was Sie mit den Schülern untersuchen wollen. Sie können unterschiedliche Primaten einbeziehen, um zu sehen, wie Menschen entstanden sind, oder Organismen aus den fünf traditionellen Reichen, um zu sehen, wie sich das Leben im Allgemeinen entwickelt hat. In der hier beschriebenen Übung werden 3 Tiere, 2 Pflanzen, 2 Algen, ein Pilz und ein Protozoon verwendet.
Sequenzen alinieren
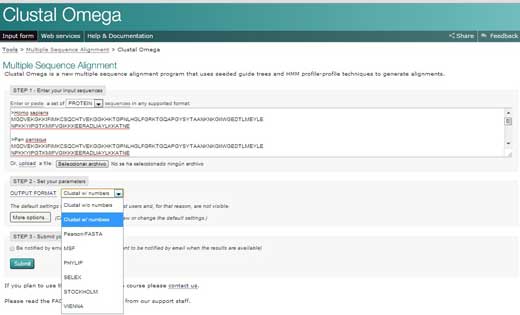
- Auf die Homepage des Europäischen Bioinformatik Institutes (EBI)w2 gehen und auf ‘Services’ klicken. Dann ‘Proteins’ aussuchen.
- Auf ‘Clustal Omega’ klicken. Den Text aus Ihrem Word Dokument in das Textfeld mit der Beschriftung ‘STEP 1’ kopieren.
- In ‘STEP 2’ ein Format für die Ausgabe des Alignments aussuchen, z.B. ‘Clustal w/ numbers’, welches die Länge jeder Sequenz anzeigen wird. Nun auf ‘Submit’ klicken, um ‘STEP 3’ zu vervollständigen.
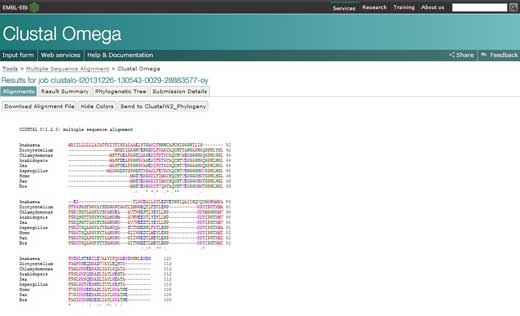
- Das Alignment für die Sequenzen wird in einem neuen Fenster erscheinen. Das erste, das man machen kann, ist auf ‘Show Colors’ zu klicken um Farben anzuzeigen. Diese Option wird jeder Aminosäure dieselbe Farbe zuordnen, sodass es einfacher ist, sie zu identifizieren.
- Um das Alignment zu analysieren, sollte man sich die folgenden Symbole merken: ein Sternchen (*) bedeutet, dass die Sequenzen an dieser Position identisch sind; ein Doppelpunkt (:) zeigt konservierte Substitutionen an (gleiche Farbe); ein Punkt (.) zeigt halb-konservierte Substitutionen an (ähnliche Struktur). Farben gruppieren die Aminosäuren nach ihren Eigenschaften. Rote sind klein, hydrophob und aromatisch; blaue sind sauer; magentafarbene sind basisch; grüne enthalten Hydroxyl-, Amin-, oder Amidgruppen oder sind basisch; alle anderen sind grau.
- Wenn man auf die Option ‘Result Summary’ klickt, kann man den Anteil identischen Positionen zwischen den unterschiedlichen Organismen nach dem Alignment sehen. In dieser Matrix kann man auch die prozentuale Identität zwischen zwei Organismen für das Protein Cytochrom C sehen. Außerdem kann man, wenn Java™ auf dem Computer installiert ist, Jalview, ein kostenloses Programm benutzen, um multiple Sequenzalignments zu editieren, visualisieren und zu analysieren. Mit Jalview kann man die Konsensussequenz für Cytochrom C, und den Grad der Konservierung für die unterschiedlichen Aminosäuren sehen.
Die Clustal Omega Software enthält viele verschiedene Optionen, die komplexere mathematische Kenntnisse voraussetzen, als es für unseren Zweck notwendig ist. Wenn Sie mehr über die Verwendung von Clustal Omega erfahren möchten, schauen Sie sich den Artikel von Sievers et al. (2011) an.
klicken.
Einen phylogenetischen Baum erstellen
- In den Clustal Omega Resultaten auf ‘Phylogenetic tree’ unten auf der Seite klicken (dazu muss Java™ installiert sein).
- Man kann sich einen phylogenetischen Baum oder ein Kladogramm ausgeben lassen. In einem Kladogramm ist die Länge der Äste willkürlich gewählt, während bei einem phylogenetischen Baum die Länge der Äste anzeigt, wie stark sich ein Protein über die Zeit weiterentwickelt hat.
Zur weiterführenden Diskussion
klicken.
- Homologe Moleküle sind ein Beispiel für divergente Evolution. Wie kann man divergente Evolution am Beispiel von Cytochrom C erklären?
- Alignments können unter Verwendung der Nukleotid- (Gen) oder der Aminosäure- (Protein) Sequenzen angefertigt werden. Warum könnte es sinnvoller sein, das Protein anstatt der DNA zu verwenden um die evolutionäre Verwandtschaft zu analysieren?
- In phylogenetischen Bäumen besteht eine ‘Klade’ aus allen Organismen, die einen gemeinsamen Vorfahren haben. Was wäre ein Beispiel für eine Klade in unserem Kladogramm?
- Welche Organismen haben sich, der phylogenetischen Analysen von Cytochrom C zufolge erst vor Kurzem zu eigenen Spezies entwickelt? Wie viele Artenbildungsereignisse gab es insgesamt?
- Warum haben sich einige Aminosäuren aufgrund von Mutationen verändert und andere nicht? Ist es möglich, dass sich konservierte Aminosäuren nicht verändert haben, weil ihre Codons überhaupt keinen Mutationen unterworfen waren?
- Markiert einige der konservierten Aminosäuren im Alignment. Untersucht ihre Funktion mit Hilfe des Internets.

Glossar
Artenbildungsereignis: Der Moment an dem sich eine Spezies in neue Spezies aufspaltet.
FASTA: Ein text-basiertes Format um Nukleotid- oder Peptidsequenzen in einem Code aus einzelnen Buchstaben darzustellen. Eine Sequenz im FASTA-Format beginnt mit einer einzeiligen Beschreibung, gefolgt von den Zeilen der Sequenzdaten. Die Beschreibungszeile wird von den anderen Zeilen durch ein Größer-als (>) Symbol unterschieden.
Homologes Protein: Homologe Proteine sind in unterschiedlichen Organismen vorhanden, die von einem gemeinsamen Vorfahren abstammen.
Kladogramm: Ein verzweigtes Diagramm, das die evolutionäre Verwandtschaft zwischen Spezies mit willkürlicher Astlänge anzeigt.
Konsensussequenz: Eine bekannte Zusammenstellung konservierter Sequenzen, oder die berechnete Reihenfolge der häufigsten Aminosäuren an jeder Position in einem Sequenzalignment.
Konservierte Aminosäure: Eine Aminosäure(sequenz) in einem Polypeptid, die sich zwischen vielen Organismen ähnelt.
Phylogenetischer Baum: Ein verzweigtes Diagramm, das die evolutionäre Verwandtschaft zwischen Spezies anzeigt, wobei die Astlänge den Unterschied zwischen den beiden Proteinen oder Genen veranschaulicht.
Danksagungen
Der Autor bedankt sich bei seiner Kollegin María Isern für ihre Hilfe mit der Englischen Grammatik im Artikel.
References
- Bromhan L. Penny D. (2003) The modern molecular clock. Nature Reviews Genetics 5: 216-224
- Kozlowski C. (2010) Bioinformatik mit Stift und Papier: wie man einen phylogenetischen Stammbaum aufstellt. Science in School 17: 28-33.
- Sievers F. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7: 539
- Dieser Artikel ist frei zugänglich auf der Homepage von Molecular Systems Biology.
Web References
- w1 – Das US National Center for Biotechnology Information bietet Zugang zu biomedizinischen und genomischen Informationen.
- w2 – Das European Bioinformatics Institute bietet freien Zugang zu Daten aus lebenswissenschaftlichen Experimenten, betreibt Grundlagenforschung in ‘Computational Biology’ und bietet ein umfangreiches Nutzter-Trainingsprogramm an, um Wissenschaftler in Akademie und Industrie zu unterstützen.
- w3 – John Kimball hat ein online Biologielehrbuch, ‘Taxonomy: Classifying Life’ (Taxonomie: die Klassifizierung des Lebens) geschrieben, welches ein Kapitel über phylogenetische Bäume ‘Phylogenetic trees’ enthält.
Resources
- Die Homepage ‘Understanding Evolution’ (Evolution verstehen) des Paläontologiemuseums der University of California bietet sehr gute Informationen über die Erstellung und Auswertung phylogenetischer Bäume.
- Hier kann man mehr über die Nutzung von Cytochrom C in phylogenetischen Bäumen lernen.
- Die Protein Data Bank (Proteindatenbank) Homepage in Europa gehört zum Europäischen Bioinformatik Institut und kann zum Suchen und Betrachten der 3D Struktur von Cytochrom C benutzt werden.
- Die Homepage Tree of Life ermöglicht es, interaktiv zu entdecken und die TV-Serie Tree of Life mit Sir David Attenborough anzusehen, die von der BBC ausgestrahlt wurde.
Review
Biologielehrer können diesen Artikel verwenden um die Themen Evolutionsbiologie, Geschichte der Wissenschaft, Biochemie und Genetik zu verknüpfen. Um möglichst viel aus diesem Artikel herauszuholen, ist es wichtig, dass Schüler die Grundlagen der DNA- und Protein-Biochemie verstehen.
Die im Artikel beschriebene Übung ist wichtig, um Schüler dazu zu motivieren, autonom in einem echten Forschungsfeld mit wissenschaftlichen Datenbanken zu arbeiten. Im Klassenzimmer können sie dazu angeleitet werden, in kleinen Gruppen Proteinsequenzen, wie die von Cytochrom C, oder DNA-Sequenzen zu vergleichen, um die Unterschiede zwischen phylogenetischen Bäumen und Kladogrammen zu erarbeiten. Bioinformatik ist sehr nützlich, um in Sekundarschulen inhalt- und sprachintegriertes Lernen auf unterschiedlichen Stufen durchzuführen, z.B. in einem interdisziplinären Projekt mit Englisch-, Geschichts- und Physiklehrern. Dieser Artikel, der diese unterschiedlichen Fächer verknüpft, könnte auch die Diskussion über den Fortschritt und die Begrenzungen solcher Forschung stimulieren.
Marina Minoli, Didaktik-Expertin, Agora Universitätszentrum, Italien