




Wykorzystanie baz danych w nauczaniu o ewolucji i biochemii Teach article
Tłumaczenie Katarzyna Badura. Narzędzia internetowe mogą zostać wykorzystane do porównania sekwencji białek i zrozumienia w jaki sposób różne organizmy ewoluowały.
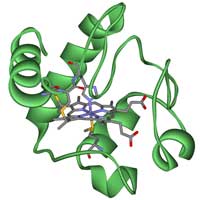
cytochromu c.
Zdjęcie dzięki uprzejmości
Klausa Hoffmeier/Wikimedia
commons
Dawniej, naukowcy prowadzili analizy ewolucyjne porównując fizyczne cechy charakterystyczne poszczególnych gatunków – znane jako fenotypy – znalezionych na wykopaliskach. Jednak wraz z odkryciem molekularnego zegara wszystko uległo zmianie.
Koncepcja zegara molekularnego wyłoniła się z obserwacji, w ramach których stwierdzono, że im więcej czasu upłynie od momentu wyodrębnienia się dwóch gatunków z jednego przodka, tym bardziej zrównicowane jest ich DNA lub sekwencje białek (w celu uzyskania bliższych informacji zobacz Bromham & Penny, 2003).
Porównując homologiczne geny lub sekwencje białek – innymi słowy te, które pochodzą z dwóch ogranizmów mających tego samego przodka – możesz zmierzyć ile czasu mineło od momentu wyodrębnienia tych organizów. Można to zobrazować na drzewie filogenetycznym.
Żeby porównać podobieństwo dwóch genów musisz ustawić odpowiednio względem siebie ich sekwencje (Kozlowski, 2010). Uzyskanie tych sekwencji było dawniej trudne, ale teraz już nie jest.
Twoi studenci zapewne mówili Ci już, że wszystko jest dostępne w Internecie – tym razem mają rację. Istnieje wiele przykładów dostępnych w Internecie bezpłatnych biologicznych baz danych, zawierających wyniki autentycznych badań, ale do tego ćwiczenia wykorzystamy głównie dwa źródła.
The National Center for Biotechnology Information (NCBI)w1 w Bethesda, MD, USA, zapewnia dostęp do bio-medycznych i genomicznych informacji, podczas gdy Europejski Instytut Bioinformatyki (European Bioinformatics Institute – EBI)w2, zlokalizowany w Hinxton, Wielka Brytania, zapewnia wolny dostęp do danych z eksperymentów przyrodniczych oraz prowadzi podstawowe badania w zakresie biologii obliczeniowej. Baza danych NCBI zapewni ci dostęp do sekwencji każdego genu lub białka, które zostały dotyczczas zsekwencjonowane. Będziesz mógł potem wykorzystać narzędzia EBI, ustawić sekwencje w odpowiednim porządku względem siebie i je przeanalizować.
Ćwiczenie
Badając ewolucyjne powiązania pomiędzy różnymi organizmami ważne jest by rozważnie wybrać gen lub proteinę, na której się skupisz. Istnieje kilka dobrze poznanych homologicznych genów, które można wykorzystać, takich jak geny białek hemoglobiny albo cytochromu c. Z czasem zajmiemy się nimi w tym ćwiczeniu. Cytochrom c jest małym białkiem, które zawiera grupę prostetyczną hem i stanowi centralny element łańcucha transportu elektronów w mitochondrium. Wszystkie organizmy tlenowe wyewoluowały od wspólnego przodka, który po raz pierwszy wykorzystał cytochrom c, jesto to więc dobry wybór zważywszy na nasz celw3.
To ćwiczenie składa się z trzech etapów:
- znalezienia sekwencji aminokwasowej cytocrhomu c w różnych organizmach,
- uporządkowania ich względem siebie i
- przygotowania drzewa filogenetycznego.
Pod koniec załączono listę pytań , które pozwolą na płynne przejście przez analizę relacji ewolucyjnych.

powiększyć
Znalezienie sekwencji białka
- Wejdź na stronę NCBIw1.
- W obszarze poszukiwań na górze strony wybierz w rozwijanej liście opcję “białko”.
- Wpisz nazwę gatunku, np. Homo sapiens i cytochrom c.
- Kliknij przycisk “szukaj”.
- Na nowej stronie pojawią się wyniki twojego wyszukiwania. Większość z nich to ta sama sekwencja z różnych źródeł, natomiast inne mogą zawierać częściowe sekwencje, lub należące do różnych gatunków lub białek. Uważnie wybierz interesujące cię białko i kliknij na link “FASTA” znajdujący się poniżej .
- Z nowej strony skopiuj ciąg liter oznaczających sekwencję aminokwasów. Wklej litery do dokumentu Word, pamiętającym o tym, by opisać sekwencję nazwą organizmu, z którego pochodzi.
- Powtórz te same czynności dla wszystkich interesujących cię organizmów, zależnie od tego co chcesz zbadać ze swoimi studentami. Możesz uwzględnić różne organizmy naczelne by zobaczyć jak ludzkość ewoluowała, lub organizmy z pięciu tradycyjnych królestw by sprawdzić, jak życie generalnie ewoluowało. W tym ćwiczeniu zostaną wykorzystane 3 zwierzęta, 2 rośliny, 2 algi, grzyby oraz pierwotniaki.
Porównanie sekwencji
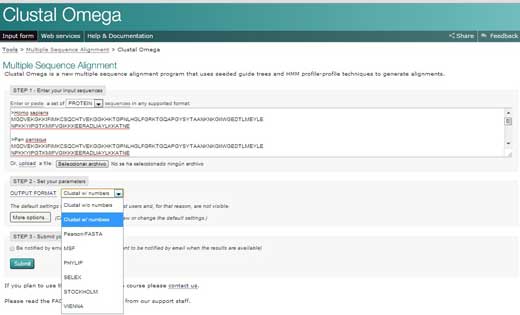
- Wejdź na stronę internetową Europejskiego Instytutu Bioinformatyki (European Bioinformatics Institute – EBI)w2 i zaznacz opcję “Services”. Następnie wybierz białka klikając “proteins”.
- Kliknij na Clustar Omega. Skopiuj tekst ze swojego dokumentu Word i wklej go w okienko oznaczone jako “STEP 1”.
- W STEP 2 wybierz format w jakim porównanie ma zostać wygenerowane, na przykład ‘Clustal w/ numbers’, które zwizualizuje długości poszczególnych sekwencji. Ostatecznie kliknij “Submit” by zrealizować ostatni krok STEP 3.
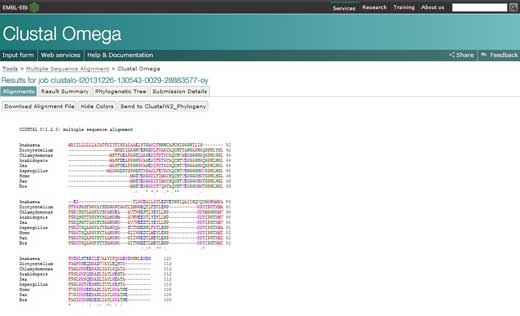
- Dopasowanie licznych sekwencji pojawi się w nowym oknie. Pierwszą rzeczą, jaką można zrobić to kliknąć by pokazać kolory. Ta opcja kolorystycznie zaznaczy takie same aminokwasy, więc ich identyfikacja będzie łatwiejsza.
- By przeanalizować dopasowanie należy zapamiętać następujące symbole: gwiazdka (*) oznacza, że sekwencje w danym miejscu są identyczne, dwukropek (:) wskazuje na konserwatywne substytucje (grupy w tym samym kolorze); natomiast kropka (.) odpowiada substytucji semikonserwatywnej (podobne kształty). Aminokwasy są grupowane kolorystycznie w zależności od cech charakterystycznych. Czerwone są małe, hydrofobowe, aromatyczne; niebieskie są kwasowe, fioletowe – proste, zielone to aminokwasy hydroksylowe, aminowe, amidowe i proste, a szare to cała reszta.
- Wybierając opcję “Result Summary” (podsumowanie wyników) będzie można poznać procent podobieństwa stwierdzonego między organizmami po wykonaniu dopasowania. W tej macierzy można poznać procent podobieństwa dwóch organizmów pod względem sekwencji białka cytochromu c. Co więcej, jeśli masz zainstalowane na swoim komputerze oprogramowanie Java™, możesz wykorzystać Jalview, darmowy program umozliwiający edycję, wizualizację i analizę dopasowanych sekwencji. Korzystając z Jalview będzie można zobaczyć zgodność sekwencji dla cytochromu c oraz poziom konserwatywności dla różnych aminokwasów.
Oprogramowanie Clustal Omega zawiera wiele opcji, które wymagają bardziej wyszukanej wiedzy matematycznej niż ta, która potrzebna jest do naszego zadania. Żeby zdobyć więcej informacji na temat wykorzystania Clustal Omega zajrzyj do artykułu autorstwa Sievers et al. (2011)
powiększyć
Przygotowanie drzewa filogenetycznego
- W programie Clustal Omega, w oknie wyników, naciśnij na “Phylogenetic tree” (Drzewo filogenetyczne) na dole (musisz mieć zainstalowane oprogramowanie Java™).
- Możesz otrzymać drzewo filogenetyczne lub kladogram. W kladogramie długość gałęzi drzew jest arbitralna, natomiast w drzewie filogenetycznym długość gałęzi wskazuje jak wiele białek ewoluowało wraz z biegiem czasu.
Pytania do dyskusji
powiększyć
- Homologiczne molekuły są przykładem ewolucji rozbieżnej. Jak możesz wytłumaczyć ewolucję rozbieżną na przykładzie cytochromu c?
- Dopasowania można wykonać wykorzystując sekwencje nukleotydów (genów) lub aminokwasów (białek). Jak uważasz, dlaczego bardziej przydatne do analizy powiązać ewolucyjnych są białka niż DNA?
- W drzewach filogenetycznych “klad” formowany jest przez organizmy posiadajace jednego przodka. Wymień podobny przykład ze swojego kladogramu.
- Biorąc pod uwagę filogenetyczną analizę cytochromu c, jakie organizmy doznały ewolucji gatunkowej ostatnimi czasy? Jaka jest całkowita suma zmian ewolucyjnych?
- Jak myślisz, dlaczego część aminokwasów uległa zmianom w wyniku mutacji, a część nie? Czy uważasz, że konserwatywne aminokwasy nie uległy zmianie dlatego, że ich kodony nie uległy żadnym mutacjom?
- Wskaż kilka z takich konserwatywnych aminokwasów w swoim dopasowaniu. Poszukaj w internecie jakie pełnią funkcje.

Słowniczek
Kladogram: Rozgałęziony diagram, o gałęziach arbitralnej długości, ukazujący ewolucyjne pokrewieństwo między gatunkami.
Sekwencja konsensusowa: Znany zestaw konserwatywnych sekwencji bądź też wyznaczona kolejność najczęściej występujących aminokwasów znalezionych na poszczególnych pozycjach w dopasowaniu.
Konserwatywne aminokwasy: Sekwencja aminokwasów w polipeptydzie, podobna w licznych organizmach.
FASTA: format tekstowy wykorzystywany do zapisu sekwencji nukleotydowych lub białkowych z wykorzystaniem kodu pojedynczych liter. Sekwencja w formacie FASTA zaczyna się od jednoliniowego opisu, po którym następuje linia danych sekwencyjnych. Opis linii odgrodzony jest od sekwencji danych symbolem większy-niż (>).
Białko homologiczne: Te białka, obecne u niektórych organizmów, które pochodzą od wspólnego przodka.
Drzewo filogenetyczne: Rozgałęziony diagram obrazujący ewolucyjne powiązania między gatunkami. Długość gałęzi każdorazowo wskazuje na różnicę pomiędzy dwoma białkami lub genami.
Ewolucja gatunkowa: Moment, w którym gatunek przodka ewoluuje w dwa nowe gatunki
Podziękowanie
Autor chciałby podziękować swojej koleżance Maríi Isern za pomoc przy sprawdzaniu angielskiej gramatyki w artykule.
References
- Bromhan L. Penny D. (2003) The modern molecular clock. Nature Reviews Genetics 5: 216-224
- Kozlowski C. (2010) Bioinformatyka z kartką i długopisem: jak zbudować drzewo filogenetyczne. Science in School 17.
- Sievers F. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7: 539
- Artykuł jest dostępny za darmo na stronie internetowej Molecular Systems Biology.
Web References
- w1 – US National Center for Biotechnology Information zapewnia dostęp do informacji biomedycznej i genomowej.
- w2 – Europejski Instytut Bioinformatyki (The European Bioinformatics Institute) zapewnia dostęp do danych z eksperymentów przyrodniczych, prowadzi podstawoewe badania w zakrecie biologii obliczeniowej i oferuje szeroki program treningowy, wsperający użytkowników w środowisku akademickim i przemysłowym.
- w3 – John Kimball napisał dostępną online książkę o tematyce biologicznej, zatytułowaną “ Taxonomy: Classifying Life”, która zawiera rodział na temat ‘Phylogenetic trees’ (rozdział dostępny w języku angielskim).
Resources
- Strona internetowa ‘Understanding Evolution’ Muzeum Paleontologii Uniwersytetu Californii ( University of California’s Museum of Paleontology) oferuje szeroki zestaw informacji w temacie budowy i sposobu czytania drzewa filogenetycznego.
- Żeby dowiedzieć się więcej o cytochromie c w drzewie filogenetycznym.
- Strona internetowa Protein Data Bank w Europie należy do Europejskiego Instytutu Bioinformatyki i może być wykorzystywana do szukania i obrazowania struktury cytochromu c w formie 3D.
- Strona internetowa Tree of Life pozwala na interaktywne odkrycie oraz oglądanie serialu telewizyjnego Tree of Life emitowanego na BBC i prowadzonego przez Sir Davida Attenborough.
Review
Nauczyciele bilogii mogą wykorzystać ten artykuł do połączenia tematów biologii ewolucyjnej, historii nauki, biochemii i genetyki. Żeby wyciągnąć z tego artykułu jak najwięcej ważne jest, aby studenci rozumieli podstawy DNA i biochemii białek.
Ćwiczenie opisane w tym artykule jest ważne w przypadku motywacji studentów do niezależnego działania w rzeczywistych badaniach z wykorzystaniem naukowych baz danych. W czasie pracy w klasie, studenci mogą zostać podzieleni na małe grupki, w obrębie których będą pracować, porównując sekwencje białek, takich jak cytochrom c, lub DNA, w celu zrozumienia różnic pomiędzy drzewem filogenetycznym a kladogramem. Bioinformatyka jest bardzo przydatna w szkole średniej do realizacji treści oraz języka ze zintegrowanym kształceniem na różnych poziomach, angażując nauczycieli angielskiego, historii i fizyki w interdyscyplinarnym projekcie. Ten artykuł, łączący w sobie trzy różne dziedziny, może również pełnić funkcję iskry rozpoczynającej dyskusję o postępowach i ograniczeniach w takich badaniach.
Marina Minoli, Expert Dydaktyczny Agora University Centre, Włochy