




O uso de bases de dados biológicas para ensinar evolução e bioquímica Teach article
Traduzido por Rita Campos. Ferramentas disponíveis na internet podem ser usadas para comparar sequências de proteínas e compreender a evolução de diferentes organismos.
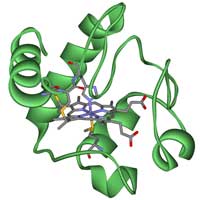
citocromo c.
Imagem cortesia de Klaus
Hoffmeier/Wikimedia
commons
No passado, os cientistas faziam análises evolutivas comparando as características físicas – designadas por fenótipos – das espécies encontradas no registo fóssil. No entanto, desde a descoberta dos relógios moleculares que este cenário mudou.
O conceito de relógio molecular surgiu por se ter observado que quanto mais tempo decorreu desde que duas espécies divergiram de um ancestral comum mais diferente será a sequência do seu ADN ou proteína (para uma revisão, ver Bromham & Penny, 2003).
Comparando sequências de genes ou proteínas homólogos – noutras palavras, sequências de dois organismos com um ancestral comum directo – pode-se medir o tempo que passou desde que os organismos divergiram. E isso pode ser visualizado numa árvore filogenética.
Para verificar quão similar dois genes são, é preciso ter as suas sequências e alinhá-las correctamente (Kozlowski, 2010). Conseguir essas sequências costumava ser muito difícil mas não agora.
Os seus estudantes provavelmente já lhe disseram que está tudo na internet – desta vez, eles estão certos. Há muitos exemplos de bases de dados biológicas de acesso livre, e que contêm dados de investigações reais, na Internet mas para esta actividade vamos usar dois recursos em particular.
O Centro Nacional para a Informação Biotecnológica (National Center for Biotechnology Information; NCBI)w1 em Bethesda, MD, EUA, oferece o acesso a informação biomédica e genómica, enquanto que o Instituto Europeu de Bioinformática (European Bioinformatics Institute; EBI)w2, localizado nem Hinxton, Reino Unido, oferece dados de experiências em ciências da vida e realiza investigação básica em biologia computacional. A base de dados do NCBI vai oferecer-lhe a sequência de qualquer gene ou proteína que já tenha sido sequenciada e depois pode usar ferramentas do EBI para alinhar e analisar essas sequências.
Actividade
Quando se investigam as relações evolutivas entre diferentes organismos é importante escolher cuidadosamente que gene ou proteína vamos usar. Há exemplos bem conhecidos de genes homólogos que podem ser usados, como os genes que codificam as proteínas hemoglobina ou citocromo b, e nesta actividade vamos usar este último. O citocromo c é uma pequena proteína heme que é o componente central na cadeia de transporte de electrões na mitocôndria. Todos os organismos aeróbios evoluíram de um ancestral comum que usou o citocromo c pela primeira vez, por isso, para o nosso propósito, é uma boa escolhaw3.
Esta actividade desenvolve-se em três secções diferentes:
- encontrar as sequências de aminoácidos do citocromo c em diferentes organismos,
- alinhá-los, e
- construir uma árvore filogenética.
Por fim, incluem-se algumas questões para guiar a investigação sobre relações evolutivas.

ampliar.
Encontrar as sequências proteicas
- Ir para a página web do NCBIw1.
- Na área de procura (search), no topo da página, seleccionar ‘proteína’ (‘protein’) do menu
- Escrever o nome da espécie, por ex. Homo sapiens, e citocromo c.
- Carregar no botão de procura (search).
- Uma nova página vai apresentar uma lista com os resultados da sua procura. A maior parte deles são a mesma sequência proveniente de diferentes fontes mas outros podem ser sequências parciais ou sequências de espécies ou proteínas diferentes. Escolher cuidadosamente a proteína de interesse e carregar na ligação que se encontra por baixo, designada ‘FASTA’.
- Da nova página que se carregou, copiar a cadeia de letras maiúsculas correspondente à sequência de aminoácidos. Colar as letras num documento Word não esquecendo de assinalar a sequência com o nome do organismo de onde vem.
- Fazer o mesmo para o número de organismos que se pretenda, dependendo do que se quer investigar com os alunos. Pode querer incluir diferentes primatas para analisar a evolução humana ou organismos dos cinco reinos tradicionais para analisar de que forma a vida em geral evoluiu. Nesta actividade vamos usar 3 animais, 2 plantas, 2 algas, um fungo e um protozoário.
Alinhar as sequências
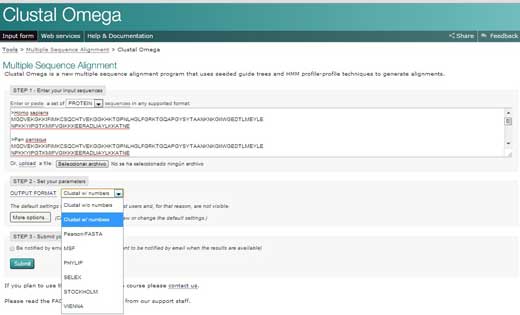
- Ir para o sítio web do EBIw2 e carregar em ‘Serviços’ (‘Services’). A seguir, escolher ‘proteínas’ (‘proteins’).
- Carregar em Clustal Omega. Copiar o texto do documento Word e colar na caixa de texto designada ‘PASSO 1’ (‘STEP 1’).
- Em PASSO 2 (STEP 2), escolher um formato para o alinhamento final, tal como ‘Clustal w/números’, que vai mostrar o tamanho de cada sequência. Por fim, carrear em ‘Submeter’ (‘Submit’) para completar o PASSO 3 (STEP 3).
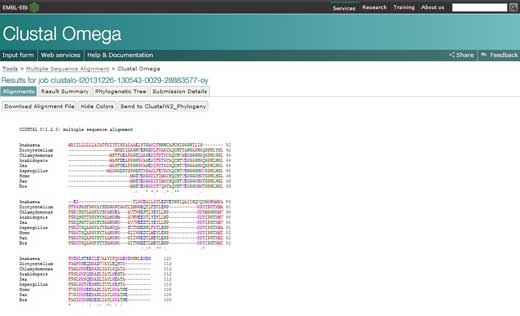
- O alinhamento das múltiplas sequências vai aparecer numa nova janela. A primeira coisa que se pode fazer é carregar no botão para mostrar as cores (‘show colours’). Esta opção dá a cada aminoácido a mesma cor e por isso é fácil identificá-los.
- Para analisar o alinhamento, manter os seguintes símbolos na cabeça: um asterisco (*) significa que as sequências são idênticas naquela posição; dois pontos (:) indicam substituições conservativas (grupo da mesma cor); um ponto final (.) refere-se a substituições semi-conservativas (formas semelhantes). As cores agrupam os aminoácidos por características: vermelho para os aminoácidos pequenos, hidrofóbicos e aromáticos; azul para os ácidos; magenta para os básicos; verde para os hidróxil, amino, amido, básico; cinzento para os restantes.
- Se carregar na opção ‘Sumários dos Resultados’ (‘Result Summary’), pode ver a percentagem de identidade conservada entre os diferentes organismos após o alinhamento. Nessa matriz encontra a percentagem de identidade de dois organismos para a sequência da proteína citocromo c. Adicionalmente, se tem o programa Java™ instalado no computador, pode usar o Jalview, um programa gratuito para editar, visualizar e analisar alinhamentos de múltiplas sequências. Com o Jalview será capaz de ver a sequência consensual para o citocromo c e o nível de conservação dos diferentes aminoácidos.
O software Clustal Omega tem muitas opções diferentes que envolvem um conhecimento matemático mais sofisticado do que o que é necessário para o propósito desta actividade. Se quiser saber mais sobre o Clustal Omega consulte o artigo de Sievers et al. (2011).
ampliar.
Fazer uma árvore Filogenética
- Nos resultados do Clustal Omega, carregue no botão ‘Árvore filogenética’ (‘Phylogenetic tree’), localizado no fundo (vai precisar de ter o Java™ instalado).
- Pode obter uma árvore filogenética ou cladística. Num cladograma o comprimento dos ramos na árvore é arbitrário enquanto numa árvore filogenética o comprimento dos ramos indicam o quanto a proteína evoluiu ao longo do tempo.
Para uma discussão mais aprofundada
ampliar.
- As moléculas homólogas são um exemplo de evolução divergente. Como pode explicar a evolução divergente usando o citocromo c?
- Os alinhamentos podem ser construídos usando sequências de nucleótidos (genes) ou de aminoácidos (proteínas). Porque será mais útil usar proteínas e não ADN para analisar relações evolutivas?
- Numa árvore filogenética, um ‘clado’ é formado por todos os organismos que têm um ancestral comum mais recente. Dê um exemplo a partir do seu cladograma.
- Que organismos sofreram um evento de especiação mais recentemente, de acordo com a análise filogenética do citocromo c? Qual é o número total de eventos de especiação?
- Pensa que alguns aminoácidos terão mudado graças à mutação mas outros não; porquê? Pensa que os aminoácidos conservados não mudaram porque os seus codões não sofreram qualquer mutação?
- Mostre alguns desses aminoácidos conservados no alinhamento. Investigue a sua função na Internet.

Glossário
Cladograma: Um diagrama ramificado que mostra as relações evolutivas entre espécies, no qual o comprimento dos ramos é arbitrário.
Sequência consenso: Um conjunto conhecido de sequências conservadas ou a ordem calculada dos aminoácidos que mais vezes se encontram em cada posição num alinhamento de sequências.
Aminoácido conservado: Uma sequência de aminoácidos num polipéptido que é semelhantes em múltiplos organismos.
FASTA: um formato de texto usado para representar sequências nucleotídicas ou peptídicas usando um código de uma letra. Uma sequência no formato FASTA começa com uma descrição em linha única, seguida de linhas de dados de sequências. A linha da descrição distingue-se das linhas das sequências por um símbolo maior-que (>).
Proteína homóloga: Aquelas proteínas partilhadas por alguns organismos que derivam de um ancestral comum directo.
Árvore filogenética: Um diagrama ramificado que mostra as relações evolutivas entre espécies, no qual o comprimento dos ramos indica a diferença entre duas proteínas ou genes.
Evento de especiação: O momento no qual uma espécie ancestral diverge em duas novas espécies.
Agradecimentos
O autor gostaria de agradecer à sua colega María Isern pela ajuda na revisão da gramática do inglês deste artigo.
References
- Bromhan L. Penny D. (2003) The modern molecular clock. Nature Reviews Genetics 5: 216-224
- Kozlowski C. (2010) Bioinformatics with pen and paper: building a phylogenetic tree. Science in School 17: 28-33.
- Sievers F. et al. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7: 539
- Este artigo está disponível gratuitamente no sítio web da revista científica Molecular Systems Biology.
Web References
- w1 – O Centro Nacional para a Informação Biotecnológica oferece acesso a informação biomédica e genómica.
- w2 – O Instituto Europeu de Bioinformática oferece gratuitamente dados de experiências nas ciências da vida, realiza investigação básica em biologia computacional e oferece um extenso programa de formação para utilizadores, apoiando investigadores na academia e na indústria.
- w3 – John Kimball escreveu um livro digital de biologia chamado ‘Taxonomia: Classificando a Vida’ (‘Taxonomy: Classifying Life’) que inclui um capítulo sobre ‘Árvores filogenéticas’ (‘Phylogenetic trees’) [sem edição portuguesa].
Resources
- O sítio web ‘Compreender Evolução’ (‘Understanding Evolution’ website) do Museu de Paleontologia da Universidade da Califórnia oferece informação muito boa sobre construir e ler árvores filogenéticas. (em português, pode ler os separadores ‘Concepções erradas sobre evolução’ e ‘Glossário’ no sítio web ‘Um livro sobre evolução’ – http://umlivrosobreevolucao.blogspot.pt/)
- Para aprender mais sobre o uso do citocromo c em árvores filogenéticas.
- O sítio web Banco de Dados Proteico (Protein Data Bank) na Europa pertence ao Instituto Europeu de Bioinformática e pode ser usado para procurar e ver as estruturas 3D do citocromo c.
- O sítio web A Árvore da Vida (Tree of Life website) permite-lhe explorar de forma interactiva e ver a série televisiva Árvore da Vida (Tree of Life), com Sir David Attenborough, que foi emitida na BBC.
Review
Os professores de biologia podem usar este artigo para ligar tópicos de biologia evolutiva, história da ciência, bioquímica e genética. Para tirar mais partido do artigo, é importante que os alunos percebam os princípios da bioquímica do ADN e das proteínas.
A actividade descrita neste artigo é importante para motivar os alunos para trabalhar de forma autónoma numa investigação real usando bases de dados científicas. No laboratório escolar, os estudantes podem ser guiados para trabalhar em grupos pequenos, comparando sequências de proteínas, tal como o citocromo c, ou ADN, para perceber as diferenças entre árvores filogenéticas e cladísticas. A bioinformática é muito útil na escola secundária para trabalhar a ‘aprendizagem integrada de conteúdo e língua’ a diferentes níveis, com professores de inglês, história e física, num projecto interdisciplinar. Este artigo, que liga estes diferentes temas, pode ser também usado para uma discussão sobre o progresso e as limitações de uma investigação como esta.
Marina Minoli, Especialista em Didáctica, Centro Universitário de Agora, Itália