




Dal gioco alla biologia d’avanguardia: l’Intelligenza Artificiale e il problema del ripiegamento delle proteine Understand article
Come i sistemi di Intelligenza Artificiale sviluppati per vincere contro gli esseri umani nel gioco possono aiutare a svelare i segreti delle funzioni proteiche?
Le proteine si avvolgono in complesse strutture tridimensionali. Determinare queste strutture è essenziale per la comprensione di molti processi biologici, ma richiede metodi lenti e costosi. La comunità scientifica cerca da oltre 50 anni di predire il ripiegamento delle proteine con mezzi informatici, ma avanzando lentamente e con risultati limitati. Dopo aver sviluppato sistemi di Intelligenza Artificiale (IA) in grado di sconfiggere giocatori umani su tavole da gioco virtuali come Go o Brizzard’s Starcraft, l’azienda DeepMind, di proprietà di Google, ha sviluppato AlphaFold2, un sistema di IA capace di predire con accuratezza numerose strutture proteiche. Il seguente articolo spiega l’importanza di questo emozionante passo avanti.
Come sono fatte le proteine?
Le proteine sono essenziali per quasi tutti i processi biologici. Nel corpo umano ne esistono circa 20 000, nel mondo diversi milioni, e ciascuna è dotata di una struttura unica.
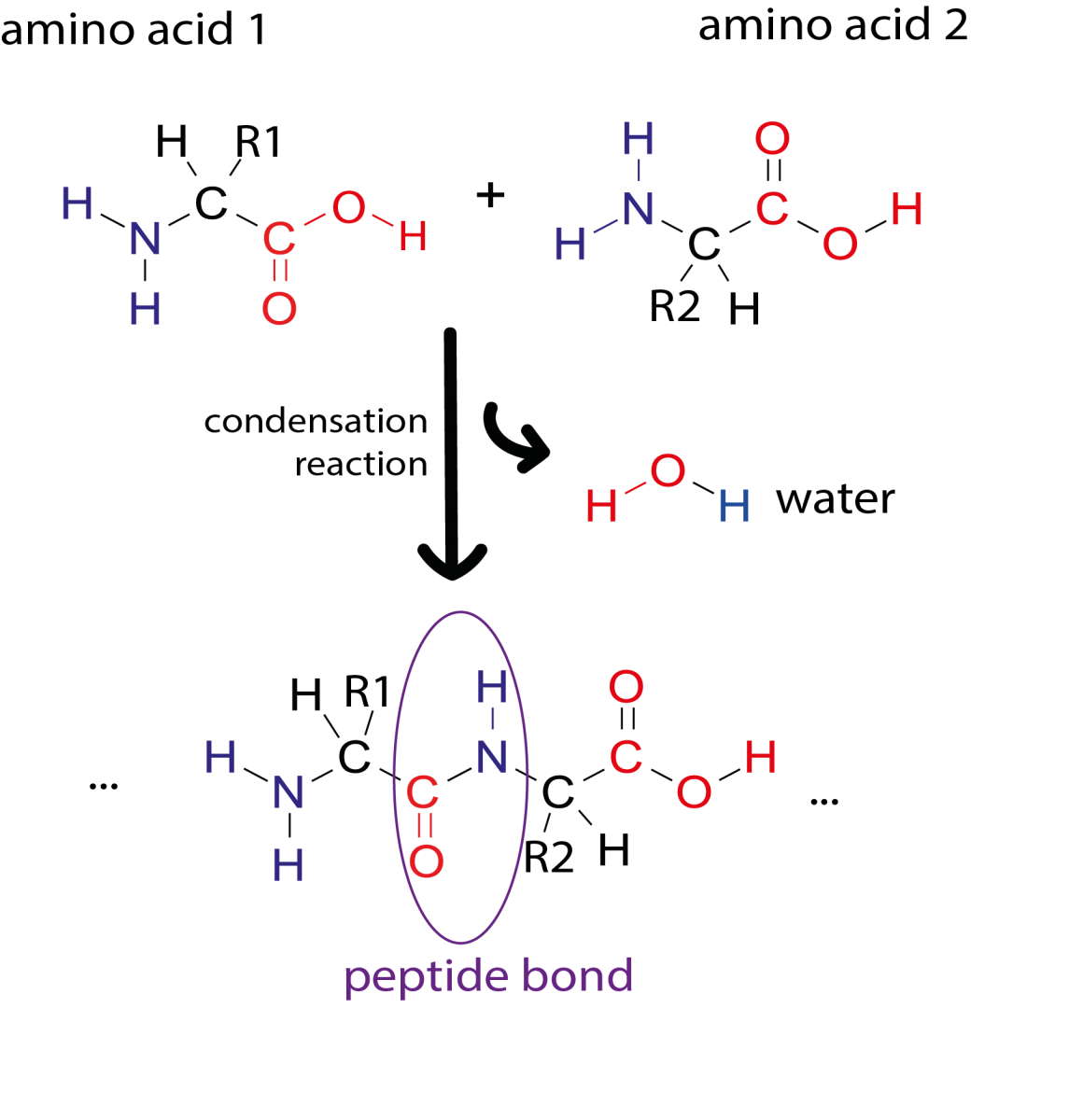
Immagine gentilmente concessa da Simone Heber
La struttura primaria di una proteina consiste nella sequenza di amminoacidi uniti da legami peptidici. Le proteine sono tutte costruite partendo da 20 amminoacidi e la loro sequenza è codificata nel DNA. Porzioni di una catena di amminoacidi possono ripiegarsi in strutture secondarie, come α-eliche e foglietti-β. Queste strutture secondarie possono poi interagire l’una con l’altra e generare forme tridimensionali complesse (struttura terziaria). Per il modo in cui una catena proteica è ripiegata su se stessa, amminoacidi che sono distanti nella struttura primaria possono trovarsi vicini nella struttura tridimensionale.
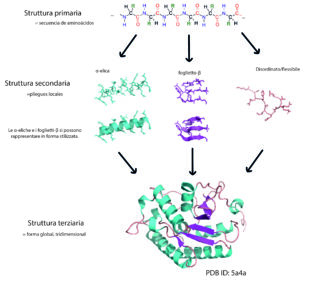
Immagine gentilmente concessa da Simone Heber
La struttura tridimensionale di una proteina determina la sua funzione e un cattivo ripiegamento può portare a malfunzionamenti e malattie. Il morbo di Alzheimer e il morbo di Parkinson sono malattie dovute a proteine mal ripiegate.
Risolvere le strutture proteiche è essenziale per la comprensione di funzioni biologiche fondamentali e può essere utile per affrontare certe malattie. Per esempio, in base alla struttura di una proteina legata a una malattia si possono progettare farmaci efficaci.
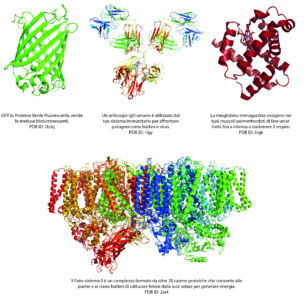
Immagine gentilmente concessa da Simone Heber
Metodi sperimentali per determinare le strutture proteiche
Nel 1962, Max Perutz e John Kendrew (che in seguito fu tra i fondatori dell’EMBL nonché il suo primo direttore) ottennero il Premio Nobel per la Chimica per aver determinato la prima struttura proteica, quella della mioglobina, tramite cristallografia a raggi X.[1]
Questo metodo impiega raggi X molto intensi, prodotti per esempio da un sincrotrone (ma alcune università hanno anche strumenti più piccoli e meno potenti), diretti su cristalli proteici. I cristalli diffrangono i raggi X e il profilo di diffrazione corrispondente consente all’osservatore di calcolare la struttura della proteina.

Christian Hendrich, GNU Free Documentation License, Version 1.2

Immagine gentilmente concessa da Simone Haber
Immagine gentilmente concessa da Simone Haber
Un secondo metodo è la spettroscopia a risonanza magnetica nucleare (NMR), che misura i campi magnetici dei nuclei atomici. Tali campi magnetici sono influenzati dalle zone in prossimità di ciascun atomo così da rivelare, nel caso degli atomi nelle proteine, quali amminoacidi si trovano vicini. Questo lavoro si è aggiudicato il Premio Nobel per la Chimica del 2002.[2]

Immagine gentilmente concessa da Simone Heber.
Un terzo metodo efficace è la microscopia crioelettronica (cryo-EM), condotta su campioni congelati, che ha meritato il Premio Nobel per la Chimica nel 2017. [3] La microscopia elettronica sfrutta un fascio di elettroni anziché di luce, permettendo la risoluzione di dettagli più minuti.
Questi metodi hanno consentito alla comunità scientifica di determinare le strutture di oltre 150 000 proteine, rese pubbliche nel Protein Data Bank (PDB).[4] Tuttavia, si tratta di tecniche lente, costose e spesso limitate dalla natura della proteina studiata. Nel tentativo di risolvere una struttura proteica, possono passare diversi anni senza alcuna garanzia di successo.
Perché non si riesce a calcolare la struttura tridimensionale di una proteina partendo dalla sua struttura primaria?
Nel 1972, Christian B. Anfinsen vinse il Premio Nobel per la Chimica per aver mostrato che la sequenza di una proteina determina la sua struttura.[5] L’idea di predire la struttura tridimensionale di una proteina dalla sua sequenza amminoacidica circola da 50 anni, e da quando si conosce la sequenza del DNA umano le strutture primarie delle proteine sono ampiamente disponibili. Allora perché finora i risultati sono stati tanto infruttuosi?
Una catena di amminoacidi può in teoria ripiegarsi in un numero enorme di strutture terziarie: per una proteina di 100 amminoacidi, si stimano 10300 (cioè 10 seguito da 299 zeri!) strutture possibili. In natura, le proteine si ripiegano generalmente nella struttura più stabile. Questa struttura di minima energia può essere calcolata, ma il confronto tra tutte le possibili strutture richiede una potenza di calcolo gigantesca. Uno dei tentativi di risolvere il “problema del ripiegamento” è il progetto di calcolo distribuito Folding@home,[6] oggi uno dei sistemi più veloci al mondo grazie al contributo di volontari in termini di potenza di calcolo. Chiunque può prendervi parte mettendo a disposizione per il ripiegamento delle proteine la capacità di calcolo inutilizzata dal proprio computer, smartphone o PlayStation3!
Il concorso CASP e il vincitore del 2020, AlphaFold
Il consorzio per l’Analisi Critica della Predizione della Struttura proteica (in inglese, CASP), fondato nel 1994[7] ogni due anni organizza un concorso in cui specifici programmi sono messi alla prova per predire strutture proteiche già risolte sperimentalmente ma non ancora pubblicate. Tuttavia, nessuno è ancora riuscito a predire precisamente la struttura di una proteina.
Nel 2018, DeepMind[8,9] ha partecipato e vinto con AlphaFold, un sistema di IA. Nell’edizione del 2020, AlphaFold2 ha segnato un ulteriore balzo in avanti riuscendo a predire con precisione sperimentale più del 90% delle strutture proteiche, superando di gran lunga gli altri concorrenti.[9–11]
DeepMind è un’azienda di proprietà di Google conosciuta per i sistemi di IA capaci di sconfiggere giocatori umani a scacchi, Go o StarCraft. Nel 2017, l’IA AlphaGo ha vinto contro il campione mondiale di Go. L’IA è stato allora riprogrammato e ha imparato a giocare a scacchi senza intervento umano.
AlphaFold2 è un’IA basata sul deep-learning: come addestramento, al sistema sono state fornite oltre 100 000 strutture proteiche note, frutto del lavoro di centinaia di ricercatori e ricercatrici. Il sistema ha poi usato gli schemi appresi dall’addestramento per predire strutture proteiche precise in pochi giorni.
Qual è l’impatto per la scienza e la società?
DeepMind intende usare AlphaFold per risolvere le strutture di proteine coinvolte in malattie umane e facilitare l’ideazione di farmaci. I dati strutturali possono anche aiutare la progettazione di enzimi che degradano la plastica o producono biocarburante. Risolvere le strutture proteiche sperimentalmente è un processo lungo e costoso: la possibilità di predirle potrebbe velocizzare sensibilmente la ricerca e abbassare i costi.
Immagine gentilmente concessa da Simone Heber
Restano tuttavia delle questioni aperte, perché sebbene i metodi basati sull’apprendimento automatico riescano a prevedere le strutture, essi non spiegano come si ripiegano le proteine. Se una proteina passasse per tutte le 10300 strutture possibili per ripiegarsi, impiegherebbe un tempo maggiore dell’età dell’universo; eppure, in natura, le proteine si ripiegano nel giro di millisecondi. A questo proposito si parla di “paradosso di Levinthal”, da Cyrus Levinthal che lo enunciò nel 1969.
Inoltre, le strutture proteiche hanno una certa flessibilità, per esempio nel legarsi ad altre proteine o a un farmaco, per cui il loro ripiegamento non dipende solo dalla sequenza primaria. Ciò significa che anche potendo predire precisamente la struttura, resterebbero comunque importanti la sua determinazione sperimentale e un’analisi funzionale.
In ogni caso, la predizione precisa della struttura è in grado di accelerare il progresso scientifico ed evitare una buon numero di esperimenti seccanti e costosi. Le predizioni possono impostare l’ideazione di esperimenti, snellire il processo scientifico e permettere alla scienza di occuparsi più rapidamente di problemi più avanzati.
References
[1] Discorso per il Premio Nobel per la Chimica del 1962, lettura veloce: https://www.nobelprize.org/prizes/chemistry/1962/speedread/
[2] Discorso per il Premio Nobel per la Chimica del 2002, comunicato stampa: https://www.nobelprize.org/prizes/chemistry/2002/press-release/
[3] Discorso per il Premio Nobel per la Chimica del 2017, comunicato stampa: https://www.nobelprize.org/prizes/chemistry/2017/press-release/
[4] Pagina principale del PDB: http://www.rcsb.org/
[5] Discorso per il Premio Nobel per la Chimica del 1972, comunicato stampa: https://www.nobelprize.org/prizes/chemistry/1972/press-release/
[6] Pagina principale del progetto Folding@home: https://foldingathome.org/
[7] Protein Structure Prediction Center: https://predictioncenter.org/
[8] Sito di Alphafold: https://deepmind.com/research/case-studies/alphafold
[9] Notizia su AlphaFold comparsa su Nature: https://www.nature.com/articles/d41586-020-03348-4
[10] MIT technology review: https://www.technologyreview.com/2020/11/30/1012712/deepmind-protein-folding-ai-solved-biology-science-drugs-disease/
[11] Il punto di vista di un ricercatore su AlphaFold2: https://www.asbmb.org/asbmb-today/science/120520/ai-makes-huge-progress-predicting-how-proteins-fol
Resources
- Insegnare l’evoluzione e la biochimica usando le banche dati biologiche online: Tenorio G (2014) Usare i database biologici per insegnare l’evoluzione e la biochimica. Science in School 29:30–34.
- Scoprire come la struttura della protein verde fluorescente determina la sue proprietà di emissione: Furtado S (2009) GFP: La vita dipinta di verde. Science in School 12:19–23.
- Saperne di più sulla cristallizzazione delle proteine e l’ESRF: Cornuéjols D (2009) Biological crystals: at the interface between physics, chemistry and biology. Science in School 11:70–76.
- Saperne di più sull’archiviazione dei dati bioinformatica all’EMBL-EBI: Stroe O (2018) Bioinformatica: la nuova “stanza delle meraviglie”. Science in School 44:20–24.
- Un’animazione sulla struttura e il ripiegamento delle proteine: https://www.youtube.com/watch?v=hok2hyED9go
Author(s)
Dr Simone Heber è ricercatrice post-dottorale all’EMBL di Heidelberg, dove studia le interazioni tra proteine e RNA durante lo sviluppo degli oociti. Ha conseguito il dottorato di ricerca in un laboratorio di biologia strutturale, usando la cristallografia a raggi X e l’NMR per studiare il riconoscimento di specifici RNA da parte di una proteina neuronale, processo che contribuisce alla memoria e all’apprendimento.

Immagine concessa da Simone Heber.
Review
Il trattamento di malattie quali l’Alzheimer, il cancro e le malattie infettive dipende strettamente dall’identificazione dei bersagli molecolari per la progettazione di nuovi farmaci. Le tecnologie informatiche e l’intelligenza artificiale hanno ridotto tempi e costi necessari. Questo articolo avvicina gli studenti al cuore della collaborazione interdisciplinare nella ricerca, come mezzo per combattere più efficacemente contro le malattie.
L’articolo è utile per l’insegnamento in biologia e chimica, per introdurre l’importanza delle strutture molecolari tridimensionali, in particolare del ripiegamento della sequenza amminoacidica, e il ruolo dell’intelligenza artificiale e della bioinformatica nella scoperta dell’informazione che si nasconde nelle nostre cellule e molecole.
Jesús López Alonso, insegnante di biologia e geologia, IES Gil y Carrasco-Ponferrada, Spagna