




Natural selection at the molecular level Understand article
We know that particular genetic sequences can help us to survive in our environment – this is the basis of evolution. But demonstrating which genetic sequences are beneficial and how they help us to survive is not easy – especially in wild populations. Jarek Bryk describes some relevant recent…

iStockphoto
When humans first left Africa some 150 000 years ago, settling in the valleys of the Tigris and Euphrates, sailing between the islands of Indonesia and trekking over the Bering Strait to America, they encountered many challenges. Coming from hot, dry African savannahs, the populations had to adapt to the local conditions, and over generations their physiology and appearance changed accordingly (Harris & Meyer, 2008). People’s skin became paler after they had lived in less sunny regions (Lamason et al., 2005). Populations whose members drank milk from domesticated animals retained the ability to digest lactose into adulthood, a feature lost soon after infancy among non-milk-drinking groups (Tishkoff et al., 2007). Populations that ate starch-rich food produced more salivary amylase, the enzyme that helps to break down starch (Perry et al., 2007).
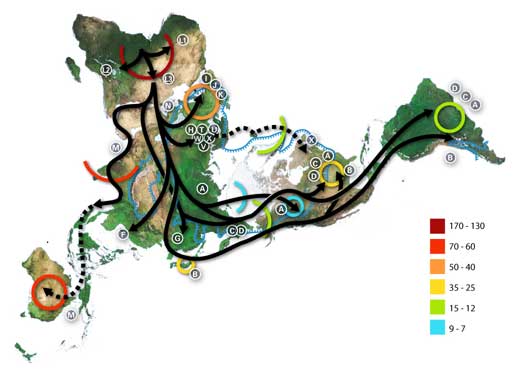
Numbers represent thousand years before present.
The blue line represents the area covered in ice or tundra during the last great ice age. The letters are the mitochondrial DNA haplogroups (pure maternal lineages); Haplogroups can be used to define genetic populations and are often geographically oriented.
For example, the following are common divisions for mtDNA haplogroups:
African: L, L1, L2, L3, L3
Near Eastern: J, N
Southern European: J, K
General European: H, V
Northern European: T, U, X
Asian: A, B, C, D, E, F, G (note: M is composed of C, D, E, and G)
Native American: A, B, C, D, and sometimes X.
Click to enlarge image
Image courtesy of Avsa; image source: Wikimedia Commons
At least some of these changes are thought to have been a consequence of positive selection (see glossary for all italicised terms). This implies that in a particular environment (the selection pressure) in the past, individuals that happened to have an advantageous DNA sequence survived and left more offspring than individuals with a different, less beneficial, sequence. Today, using the genomic sequences of many species, including humans and their closest evolutionary relatives, scientists can compare traits and DNA sequences from populations or species with different lifestyles and from different environments to identify which sequences may have played a role in the adaptations. This, in turn, allows researchers to investigate the function of a DNA sequence and its potential adaptive value for an organism.

iStockphoto
Some of the genes known to affect skin colour in humans show a specific geographic pattern of sequence variation; in particular, sequence comparisons between European and African populations suggest that variation in skin colour is due to positive selection. Lightness of skin positively correlates with increasing latitude, and several hypotheses have been proposed to explain its potentially advantageous effects.
One hypothesis, which states that light skin favours the production of vitamin D, is supported by observations that dark-skinned people living at high latitudes suffer from vitamin D deficiency. Furthermore, light skin is more sensitive to the harmful effects of sunlight: greater exposure to sunlight correlates with increased incidence of skin cancer in pale-skinned people. Therefore, pale skin in human populations living at higher latitudes may be an evolutionary compromise between protection from the carcinogenic effects of sunlight and allowing sufficient production of an essential vitamin.
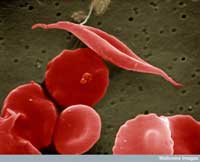
image of sickle-cell and
other red blood cells
Image courtesy of EM Unit,
UCL Medical School, Royal
Free Campus / Wellcome
Images
Although this is a solid hypothesis, the evidence behind it is indirect. A direct demonstration of the adaptive value of this trait would require measuring whether, at higher latitudes, individuals with paler skin show improved survival and reproduction. Such demonstrations in our species, however, are difficult: survival experiments (in which individuals with different traits are exposed to an environment to see which survive) cannot be performed on humans, and our long generation time makes it difficult to investigate differences in reproductive rates. The circumstances in which it is possible to observe the adaptive value of any trait in humans are therefore very limited — but they do exist.
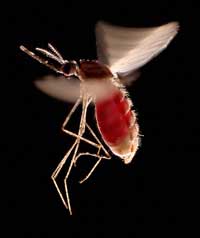
full of blood. This species,
Anopheles stephensi, is the
insect vector that transmits
malaria in India and Pakistan
Image courtesy of Hugh Sturrock
/ Wellcome Images
One example involves two diseases: sickle-cell anaemia and malaria. The gene involved in sickle-cell anaemia has two variants, or alleles: a ‘normal’ allele and a sickle-cell allele. Individuals with two sickle-cell alleles suffer from serious sickle-cell anaemia, whereas those with one sickle-cell and one normal allele do not exhibit such severe symptoms. Mortality data suggest that the sickle-cell allele can, however, be advantageous: in populations exposed to the malaria parasite, individuals carrying one sickle-cell allele and one normal allele are more likely to survive than people carrying two normal alleles, because the parasite (Plasmodium falciparum) requires healthy blood cells to invade and multiply. Therefore, the frequency of the allele that causes sickle-cell anaemia increases in malaria-exposed groups – the allele is adaptive in this environment.
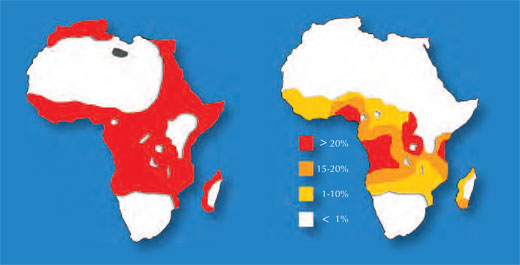
Image courtesy of Anthony Allison; image source: Wikimedia Commons
Another example demonstrating the adaptive value of a human trait concerns a fragment of chromosome 17, known to have been inverted in our ancestors more than three million years ago (Stefansson et al., 2005). The fact that this variant spread across European populations suggests that it has been positively selected for – it has conferred an advantage on individuals that carry it. By genotyping almost 30 000 Icelanders, scientists investigating the hypothesis were able to determine that over the last 80 years, individuals who carried the sequence variant had on average 3.2% more offspring per generation than individuals with the normal sequence, a plausible explanation of how the variant came to spread so rapidly.
Although the two examples clearly demonstrate the recent action of positive selection in humans, the molecular mechanisms of how the sequence variations confer their advantages are not well understood and must be investigated on a case-by-case basis. To elucidate the causal relationships between putatively adaptive DNA sequences and an individual’s fitness, scientists turn to organisms that are easier to experiment on than humans.
For instance, coat colour in the Oldfield mouse, Peromyscus polionotus, matches the soil of the habitat, providing camouflage. Mice living on the pale sands of Florida beaches are much lighter than inland-living mice of the same species. The adaptive value of this trait was demonstrated experimentally more than 30 years ago: mice with a coat that matched the soil colour were eaten less frequently by owls than the other, less camouflaged mice. However, scientists have only recently identified the genetic loci behind this adaptive trait (Hoekstra et al., 2006): variation in coat colour largely depends on different alleles of the McR1 gene. The protein encoded by this gene acts as a biochemical switch driving the production of either eumelanin, a dark pigment in the skin, or pheomelanin, a light pigment. The different alleles of the McR1 gene activate the pigment-producing pathway to a different extent, favouring the production of one pigment or the other.
micrograph of clusters of
methicillin-resistant
Staphylococcus aureus
bacteria
Image courtesy of Annie
Cavanagh / Wellcome Images
Another example of a demonstrated causal relationship involves Staphylococcus aureus, a bacterium that can cause various diseases including pneumonia or heart valve inflammation. In a rare natural experiment, a patient with recurrent S. aureus infections was treated for three months with vancomycin, one of the few antibiotics that are still effective against S. aureus. Before and at intervals throughout the treatment, scientists collected samples (isolates) of the pathogen and sequenced the entire genome of the first and last isolates. When they compared the three million base pairs (the ‘letters’ of the genetic code) that constitute this bacterium’s DNA, they found only 35 differences between the first and last isolates.
By partially sequencing the intermediate isolates, the scientists then worked out the order in which these changes must have occurred. By testing the bacterial resistance to vancomycin in vitroin the different isolates, they were able to correlate particular genetic changes with effects on the bacteria’s growth and response to the drug. For instance, the first and second isolates of bacteria differed by six nucleotide substitutions (changes to the ‘letters’) in two genes. These six mutations alone were clearly advantageous: they increased the bacterium’s tolerance to vancomycin four-fold, allowing bacteria carrying these mutations to survive and reproduce better, becoming more common in the patient’s body. Twenty-six subsequent mutations over the following weeks of treatment doubled the tolerance, effectively producing a vancomycin-tolerant strain of S. aureus (Mwangi et al., 2007).
In short, investigating the molecular basis of adaptive evolution in wild populations is not easy. The challenges include defining the selective pressures, identifying the DNA sequences behind the associated traits, measuring individuals’ fitness, and finding mechanistic explanations for how the sequence changes influence the adaptive traits. However, with the use of model organisms and recent technological developments, these investigations are now becoming feasible, increasing our understanding of how specific changes at the genetic level allow organisms to adapt to their environment.
Glossary
Adaptive value: a trait has an adaptive value if it enables an individual to survive and reproduce better in a given environment than individuals that do not possess this trait. More formally, a trait is regarded as adaptive if it increases fitness.
Allele: a variant of a gene.
Fitness: a hard-to-define formal term from evolutionary biology and population genetics; it describes the average number of offspring over one generation that are associated with one genotype compared to another genotype in a population. Thus genotypes that produce more offspring have greater fitness. For a good overview of fitness and genotype, see Wikipediaw1.
Genome: the total DNA of an organism. This is usually understood to be the nuclear DNA, as opposed to mitochondrial or plastid DNA. For further information, see ‘What is a genome’ on the US National Library of Medicine websitew2.
Positive selection: natural selection is one of the mechanisms of evolution; it describes the different survival and reproduction of individuals in a given environment. Natural selection is called ‘positive’ when it promotes certain traits that help individuals who have them, to survive and reproduce better than others.
Selection pressure: a feature of the environment (e.g. temperature; presence of parasites; predation or aggression from members of the same species) that imposes differential survival and reproduction of individuals.
Trait: one or a set of features of an organism’s characteristics (e.g. height; resistance to antibiotics; ability to see colours or to roll one’s tongue).
Acknowledgements
The author is grateful to David Hughes, Mehmet Somel and Ania Lorenc for helpful comments on the article.
References
- Harris EE, Meyer D (2006) The molecular signature of selection underlying human adaptations. American Journal of Physical Anthropology 131(S43): 89-130. doi: 10.1002/ajpa.20518
- This article provides a good overview of research into molecular evolution in humans.
- Hoekstra H et al. (2006) A single amino acid mutation contributes to adaptive beach mouse color pattern. Science 313: 101-104. doi: 10.1126/science.1126121
- This and other papers on mouse coat coloration by Hopi Hoekstra’s research group are available on the Harvard University website. See: http://www.oeb.harvard.edu/faculty/hoekstra/publications.html
- See also the follow-up paper in which the discovery of Agouti, a negative regulator of McR1 contributing to adaptive coat colour in Peromyscus, is described:
- Steiner CC, Weber JN, Hoekstra HE (2007) Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biology 5: e219. doi: 10.1371/journal.pbio.0050219
- This and all other articles in PLoS Biology are freely available online.
- The following article reviews the adaptive pigmentation of vertebrates:
- Hoekstra HE (2006) Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity 97: 222-234. doi: 10.1038/sj.hdy.6800861
This article is freely available to download from the Heredity journal website: www.nature.com/hdy
- An overview of Hopi Hoekstra’s latest research is available on John Hawks’ blog: http://johnhawks.net/weblog/topics/evolution/selection/hoekstra-2009-adaptive-pigmentation.html
- Lamason RL et al. (2005) SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310: 1782-1786. doi: 10.1126/science.1116238
- Mwangi MM et al. (2007) Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proceedings of the National Academy of Sciences of the United States of America 104: 9451-9456. doi: 10.1073/pnas.0609839104
- Perry GH et al. (2007) Diet and the evolution of human amylase gene copy number variation. Nature Genetics 39: 1256-1260. doi: 10.1038/ng2123
- See also the overview of this research at Panda’s Thumb: http://pandasthumb.org/archives/2008/12/amylase-and-hum.html
- Stefansson H et al. (2005) A common inversion under selection in Europeans. Nature Genetics 37: 129-137. doi: 10.1038/ng1508
- See also overview of the paper at Evolgen: http://evolgen.blogspot.com/2005/02/human-inversion-under-selection.html
- Tishkoff SA et al. (2006) Convergent adaptation of human lactase persistence in Africa and Europe. Nature Genetics 39: 31-40. doi: 10.1038/ng1946
- See also an overview of this research in The New York Times: www.nytimes.com/2006/12/10/science/10cnd-evolve.html?_r=1
Web References
- w1 – For a good overview of the terms ‘fitness’ and ‘genotype’, see Wikipedia: http://en.wikipedia.org/wiki/Fitness_(biology) and http://en.wikipedia.org/wiki/Genotype
- w2 – For more information about genomes and the Human Genome Project, see ‘What is a genome’ on the US National Library of Medicine website: http://ghr.nlm.nih.gov/handbook/hgp/genome
Resources
- If you found this article interesting, you may like to read some other Science in School articles about evolution:
- Haubold B (2010) Review of Why Evolution is True. Science in School 14. www.scienceinschool.org/2010/issue14/evotrue
- Leigh V (2008). Interview with Steve Jones: the threat of creationism. Science in School 9: 9-17. www.scienceinschool.org/2008/issue9/stevejones
- Patterson L (2010) Getting ahead in evolution. Science in School 14: 16-20. www.scienceinschool.org/2010/issue14/amphioxus
- Pongsophon P, Roadrangka V and Campbell A (2007) Counting Buttons: demonstrating the Hardy-Weinberg principle. Science in School 6: 30-35. www.scienceinschool.org/2007/issue6/hardyweinberg
- For more information about malaria, see:
- Hodge R (2006) Fighting malaria on a new front. Science in School 1: 72-75. www.scienceinschool.org/2006/issue1/malaria
- To learn more about the structure of starch, which salivary amylase helps to break down, see:
- Cornuéjols D (2010) Starch: a structural mystery. Science in School 14: 22-27. www.scienceinschool.org/2010/issue14/starch
Review
The article describes a range of interesting examples of evolutionary adaptations at the molecular level in humans. The difficulty in elucidating causal relationships between adaptive DNA sequences and individual fitness in humans and the need to use other experimental organisms are highlighted.
The article provides excellent material for comprehension questions focusing on the understanding of natural selection and fitness in humans and experimental organisms. For example:
- Explain the processes involved in natural selection.
- What do you understand by the term ‘fitness’?
- Explain how the sickle-cell allele confers a selective advantage in some human populations.
- What are the problems associated with establishing causal relationships between adaptive DNA sequences and fitness in humans?
- Construct a flow chart to explain the adaptive value of coat colour in Oldfield mice.
- How were the scientists able to correlate genetic changes in Staphylococcus aureus with bacterial growth and antibiotic responsiveness?
This article also enables students to research the link between DNA, amino acid sequence, protein structure and function in sickle-cell anaemia. The text is suitable for directing discussion in the classroom on the methods and problems associated with investigating the molecular basis of evolutionary relationships and the ethics of genetic testing in human populations. Interdisciplinary studies could be organised around the history of science and evolutionary population genetics.
Mary Brenan, UK