




Getting a grip on genetic diseases Understand article
Sabine Hentze and Martina Muckenthaler tell Lucy Patterson about their work – detecting genetic diseases and counselling potentially affected patients.
/ iStockphoto
As part of the recent SET-routesw1 Insight Lecturesw2 series, two scientists from Heidelberg, Germany, talked about their work and experience in the field of genetic disease: Sabine Hentze, a medical doctor specialising in human genetics and genetic counselling, and Martina Muckenthaler, a professor at the Centre for Paediatric Medicine at Heidelberg University Hospital working on the hereditary iron storage disease haemochromatosis (see box).
Genetic diseases are caused by abnormalities in a person’s DNA. These can be as simple as a single nucleotide mutation in a single gene, or as complex as deletions and rearrangements of parts of or entire chromosomes.

Muckenthaler (right)
Images courtesy of EMBL Photolab
Our DNA is constantly under attack by a range of factors that can cause mutations, such as naturally occurring radiation (for classroom experiments on radiation, see Peralta & Oliveira, 2009). In addition, copying the entire DNA sequence of a cell during each mitosis is not a fail-safe process. In response, each cell in our body keeps a fleet of DNA-repair enzymes that constantly patches up the damage. From time to time, however, mutations escape the repair process and become fixed in the DNA.
If these mutations occur in important genes, they can cause serious diseases. As we have two copies of each gene (one from our mother and one from our father), mutations that damage only one copy of the gene do not necessarily cause immediate problems, as we still have one healthy copy.
Indeed, we all carry between five and ten such recessively mutated genes without even realising it. Only dominant mutations will manifest as a disease when just one copy of the gene is damaged. Recessive mutations can become problematic, though, if both your mother and father happened to carry a mutant copy of the same gene: there is a risk that you could inherit both. This can cause diseases such as cystic fibrosis or sickle cell anaemia.
When whole sections of chromosomes go missing or are switched around, or even entire chromosomes are duplicated or deleted, these aberrations are often lethal, so that the child dies before birth, or they lead to mental retardation and malformations, syndromes such as Down syndrome.
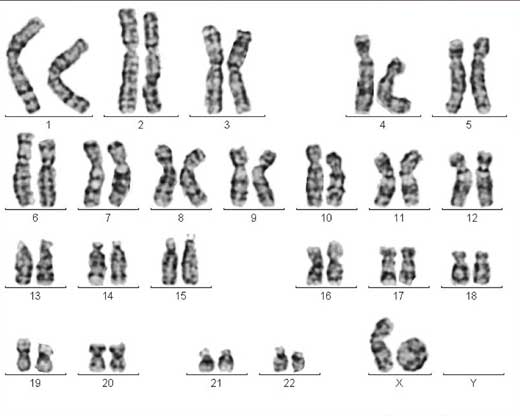
One of the X chromosomes forms
what is known as a ring
chromosome, causing Turner
syndrome. Click to enlarge
Image courtesy of Sabine Hentze
It is also possible to inherit increased susceptibility to diseases. Certain mutations, although they do not actually cause diseases, dramatically increase a person’s risk of developing particular conditions: for instance, inherited mutations in the BRCA1 and BRCA2 genes lead to an increased risk of breast cancer.
It is part of Sabine’s job to investigate and diagnose these kinds of diseases in her patients: “A typical situation is one where parents introduce me to their child, saying ‘He has not developed properly for his age. Our neighbour’s son who is the same age is much taller, he’s already crawling. Mine can’t do that’.” There are a number of tools and tests available, but when it comes to making a diagnosis, she says: “First and foremost, at the centre are we, the doctors, whose job it is to look, listen, examine and assess.” After a thorough clinical examination, and armed with a detailed family history, Sabine may already be able to guess what might be the underlying cause of a patient’s disorder. The next step is to find out what kind of mutation lies at the heart of the disease.
Chromosome analysis was first used by French geneticist and paediatrician Jérôme Lejeune, who in 1959 determined that children suffering from Down syndrome had an extra copy of chromosome 21. Even today, there are many questions that can be more fully answered using this technique than with a genetic test: cells taken from a patient are cultured in the lab, then fixed, prepared and stained so that their chromosomes can be studied under the microscope.
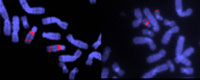
specific sequences of DNA.
Note the deletion in chromosome
22 in the sample on the right.
This causes DiGeorge Syndrome,
which is marked by the absence
of thymus and parathyroid glands,
and results in impaired immunity,
short stature, and deformities of
the face, heart and great vessels
Image courtesy of John Crolla,
Wellcome Images
Yet some diseases are caused by rearrangements or deletions too small to be seen by conventional chromosome analysis, so a technique called FISH (fluorescent in situ hybridisation) is used. Tiny, fluorescently labelled DNA probes are designed to hybridise, or stick, to a specific sequence in the region of the chromosome which is thought to be affected, and doctors check if and where they hybridise on a sample of the patient’s chromosomes.
So when do we ultimately use a genetic test? “I use one if I strongly suspect a particular disease for which we know the gene and the possible mutation,” says Sabine.
Haemochromatosis, the disease Martina has specialised in, is not only the most common hereditary disease in the western world, but in 85-90% of cases in central Europe, is caused by one specific single mutation in the HFE gene (see box below). If patients seem to be suffering from the disease or are concerned that it runs in their family, it is possible to carry out a genetic test to look for HFE mutations. The patient’s DNA is isolated, usually from a blood sample, and the nucleotides of the HFE gene in which the most common point mutations occur are sequenced, and compared with the sequence found in healthy individuals.
Haemochromatosis
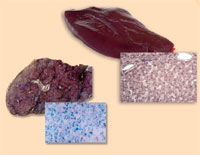
haemochromatosis (right),
compared with a healthy liver
(left). Note the excess iron
(stained with Prussian blue)
Image courtesy of Martina
Muckenthaler
Haemochromatosis is the most common hereditary disease in the western world. It is a condition that causes the body to absorb and store dangerously high levels of iron from the diet in the liver, heart, pancreas and other tissues. Besides a bronze pigmentation of the skin, this can eventually cause liver failure, heart failure or diabetes, since humans, like most animals, have no means to excrete excess iron.
Iron is an essential component of haemoglobin, the molecule responsible for oxygen transport in red blood cells. In addition, it is a cofactor of cytochromes, proteins which are important in generating energy in the respiratory chain of each cell. In the past, it was noticed that the then-common practice of bloodletting using leeches seemed to alleviate the disease. In fact, this remains the mainstay of treatment for haemochromatosis today, albeit without the leeches.
The synthesis of new red blood cells helps to use up excess iron. Initially, the disease was thought to affect only men, becoming evident only in their forties and fifties. However, we now know that women are just as likely to have the disease, but that loss of blood through menstruation and pregnancy naturally helps to alleviate the symptoms.
In 85-90% of cases in central Europe, haemochromatosis is caused by one specific mutation in the HFE gene, located on chromosome 6. It is thought to have spontaneously originated about 500 BC in one individual of a Celtic tribe living in the Danube valley, and from there it spread across Europe, and with emigrants to America and Australia. In Australia, all patients can actually be traced back to one single immigrant carrying the mutation. One in eight people in the western world carry this mutation, but since it is recessive, only one in every 250 people will develop symptoms of haemochromatosis.
Researchers like Martina Muckenthaler think that it is likely that the HFE mutation spread so widely as it conferred a selective advantage: “Historically, women gave birth to many children. Both the developing child and the births themselves, which resulted in blood loss, emptied the iron stores. Besides, there was very little meat to eat, and since this contains the most iron in our diet, it was very difficult to replenish ones iron stores. Moreover, in the past, humans tended not to live so long, so the symptoms of the disease didn’t get to develop. This means that the mutation of the HFE gene was a great advantage in terms of natural selection: people with this mutation were better able to reproduce successfully than those without the mutation.”
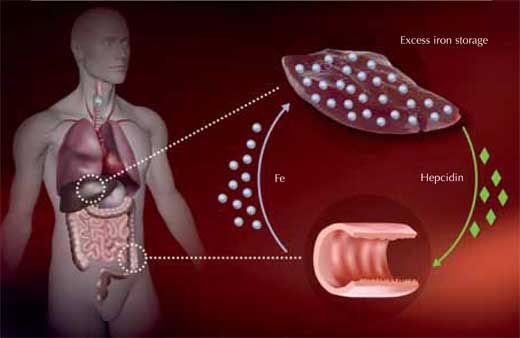
regulated in people with
haemochromatosis, leading to
excess iron being taken up in
the intestines and stored in
the liver. Click to enlarge image
Image courtesy of Martina Muckenthaler
How does this mutation cause excess iron uptake? The body absorbs iron in the intestine via a specialised transport protein. Iron is then stored inside liver cells bound to an iron storage protein, ferritin, until it needs to be mobilised. Too much iron can lead to the production of oxygen radicals, damaging cells and causing them to die.
To ensure that no surplus iron is taken up, the liver measures how much iron is available and translates this information into the production of a hormone named hepcidin – the more iron, the more hepcidin. In the intestine, hepcidin destroys the iron transporter proteins, so they won’t take up any more iron.
Martina has developed a special microarray containing 500 genes related to iron metabolism and using the mouse as a model organism. With this method, her group compared the gene activity of liver cells from healthy mice with that of liver cells from mice engineered to carry the HFE mutation that causes haemochromatosis in humans. They found that with the HFE mutation, hepcidin is not sufficiently up-regulated even when there is already enough iron in the body, causing an excess iron uptake. Through their research, Martina and others are now trying to understand this process in more detail.
When children suffer from an unspecific mental or physical disability, mutations in a range of genes on different chromosomes can be the cause. Here, chromosome-staining techniques don’t have a sufficiently high resolution to identify the defective gene – however, it would be an incredible amount of work to sequence all potentially affected genes individually. In these cases, geneticists like Sabine are starting to use a new technology: microarrays (for an in-depth explanation of microarrays and a suggestion of how to introduce them in the classroom, see Koutsos et al., 2009). Microarrays vastly speed up the process of genetic testing; as tens or hundreds of thousands of regions of the genome can be tested at the same time, it is possible to test for many disorders simultaneously. In the future, scientists hope that it might be possible to develop a microarray that can test for all genetic diseases and predispositions in one quick and simple test.
Image courtesy of ktsimage
/ iStockphoto
No parents would wish their child to be born with a genetic disease. Until recently, prenatal screening was the only option available to determine whether a baby would be born with a serious disorder. However, since the advent of in vitro fertilisation, it has become possible to examine the genetic makeup of an embryo before it is implanted into the womb, a technique called pre-implantation genetic diagnosis.
At present, many tests are available for well-known genetic diseases, allowing parents who are at risk of passing on a genetic disease to select a healthy embryo for implantation. In many respects this is great news, as it means that expectant parents do not have to go through the process of screening the embryo in the womb (amniocentesis, which itself carries a risk to the baby), waiting for the results, and taking the decision whether to terminate the pregnancy if the outcome is bad.
The more we learn about the genetic basis of different diseases and traits, and the more sophisticated our screening methods become, the more we can screen for. Of course, no parent would want their child to suffer from a serious disease – but what about less severe disorders such as haemochromatosis, congenital deafness, or even short-sightedness? Who is to say that a child growing up with such a condition wouldn’t live as full a life as an otherwise healthy person? In essence, this issue raises the subject of what is normal. Where do we draw the line as to which genetically determined conditions or traits are acceptable and which are not? Furthermore, should parents ever be allowed to choose if their child is male or female, how tall or attractive or how intelligent they are?

Another question raised by genetic testing is whether you would really want to know what your genes have in store for you. What if you were to find out that you were at high risk of developing a serious disease? You might be able to adjust your lifestyle to delay its onset or decrease the symptoms, but how would you feel if you knew your risk? And what if insurance companies or prospective employers were to have access to that information? What if there was a risk that you could pass this disease on to your children? Wouldn’t your partner want to know? Might it change the way he or she felt about you? For suggestions on how to stimulate discussions on such issues in the classroom, see Strieth et al. (2008).
It is at this point that the work of genetic counsellors like Sabine Hentze is really essential: “Besides my laboratory work, I spend much of my time on counselling patients, in other words on communication: what does this test result mean? What does it mean for me, for our child, for our family, for our future?”
And it is through the work of genetic counsellors that we have come to realise that perhaps one of the most important considerations in genetic testing is that people also have the right not to know.
References
- Koutsos A, Manaia A, Willingale-Theune J (2009) Fishing for genes: DNA microarrays in the classroom. Science in School 12: 44-49.
- Peralta L, Oliveira C (2009) Radioactivity in the classroom. Science in School 12: 57-61.
- Strieth L et al. (2008) Meet the Gene Machine: stimulating bioethical discussions at school. Science in School 9: 34-38.
Web References
- w1 – For more information about the SET-routes organisation, promoting women in science, see www.set-routes.org
- w2 – The SET-routes Insight Lectures are a series of interactive scientific lectures for use in schools. Presented by exceptional women scientists, the lectures introduce the exciting world of science, engineering and technology (SET), covering fields as diverse as space science; climate change; genetic counselling; haemochromatosis and DNA chips; malaria; stem cells and regeneration; archaeology of the Universe; and cosmology. See: www.set-routes.org/lectures
Resources
- Democs card games to debate the topics of pre-implantation diagnostics and over-the-counter genetic tests can be downloaded free here: www.neweconomics.org/gen/democs.aspx
- For a Science in School article about Democs, see: Smith K (2007) Democs: a conversation card activity for teaching science and citizenship. Science in School 4: 27-19.
- The Genes are Us website offers short films and classroom activities about genetic diseases, see: www.genesareus.org
- For an introduction to many of the most common genetic diseases, see the Genetic Disorders Library (http://learn.genetics.utah.edu/content/disorders/whataregd) section of Learn.Genetics (http://learn.genetics.utah.edu), the Genetic Science Learning Center from the University of Utah, USA.
- To learn more about oxygen radicals in your body and how to counteract them, see: Farusi G (2009) Farusi G (2009) Looking for antioxidant food. Science in School 13: 39-43.
- If this article has whet your appetite, you can find further reading on the most cutting-edge advances and opinion on genetic testing and personal genomics at Daniel MacArthur’s science blog, Genetic Future: http://scienceblogs.com/geneticfuture
Review
Inherited diseases and syndromes are pivotal for any genetics class: the topic is relevant to all students and will encourage them to involve the family in ‘family-tree research’. It will trigger even more discussions in the class if students are ready to talk about rare diseases or chromosome aberrations in their extended family. Haemochromatosis is an issue that need not be avoided, since it is medically manageable when diagnosed. It is also a good example of an evolutionary advantage that can lead to a dead end when the environment changes. This promises to spark many interesting discussions.
In addition, the article describes all major techniques that are currently used to analyse inherited defects, including a cutting-edge technique, the microarray.
Possible topics for discussion include: genetic ethics in general; in vitro fertilisation; pre-implantation genetics; family planning in cases of known diseases; and the question of when life starts. All these are topics related to ethics and religion.
Here are a few suggestions for tasks to set students, using this article:
- In a table, list the described techniques and their feasibility in determining an inherited syndrome or disease.
- Discuss the features of each technique concerning its validity for the patient.
- Discuss ‘the right to know’ or ‘not to know’ and the ethical dilemma that doctors face when they discover severe genetic mutations.
- Illustrate iron uptake and regulation in humans using a flowchart or pictures.
- Draw a diagram of the inheritance pattern of the mutation in the HFE gene, using Mendelian genetics.
Friedlinde Krotscheck, Austria