




Oyundan son teknoloji biyolojiye: Yapay zeka ve protein katlanma sorunu Understand article
Oyunlarda insanları yenmek için geliştirilen yapay zeka sistemleri gibi, yapay zeka protein işlevinin sırlarını çözmeye nasıl yardımcı olabilir?
Proteinler karmaşık 3D yapılara katlanır. Bu yapıları belirlemek, birçok biyolojik süreci anlamanın anahtarıdır, ancak zaman alıcı ve pahalı deneyler gerektirir. Bilim adamları yaklaşık 50 yıldır protein kıvrımlarını hesaplamalı olarak tahmin etmeye çalışıyorlar, ancak ilerleme yavaş ve başarı sınırlıydı. Google’ın sahip olduğu DeepMind şirketi, soyut masa oyunu Go veya Blizzard’ın Starcraft’ında insanları yenen yapay zeka (AI) sistemleri geliştirdikten sonra, kısa süre önce birçok protein yapısını deneysel doğrulukla tahmin edebilen bir AI sistemi olan AlphaFold2’yi geliştirdi. Aşağıdaki makale bu heyecan verici atılımın önemini açıklamaktadır.
Proteinler neye benziyor?
Proteinler hemen hemen tüm biyolojik süreçlerin anahtarıdır. İnsan vücudunda yaklaşık 20.000, yeryüzünde ise milyonlarca farklı protein vardır ve her biri kendine özgü bir yapıya sahiptir.
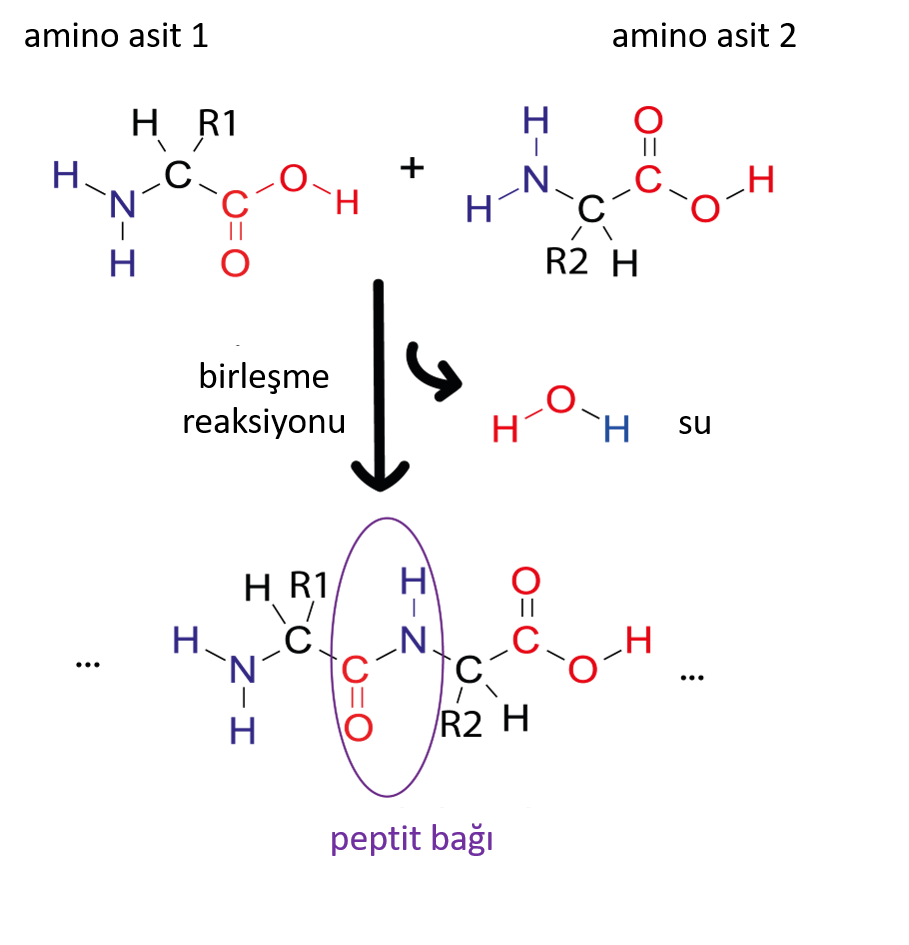
Resim Simone Heber tarafından sağlanmıştır
Bir proteinin birincil yapısı, peptit bağlarıyla birbirine bağlanan bir dizi amino asitten oluşur. Proteinler 20 farklı amino asitten oluşur ve dizilimleri DNA’mızda kodludur. Bir amino asit zincirinin bölümleri, α-sarmallar ve β-tabakaları gibi ikincil yapılara katlanabilir. Bu ikincil yapılar daha sonra karmaşık 3D şekiller (üçüncül yapı) oluşturmak için birbirleriyle etkileşime girebilir. Protein zincirinin kendi etrafında katlanma şekli nedeniyle, birincil yapıda birbirinden uzak olan amino asitler 3 boyutlu yapıda birbirine yakın olabilir.
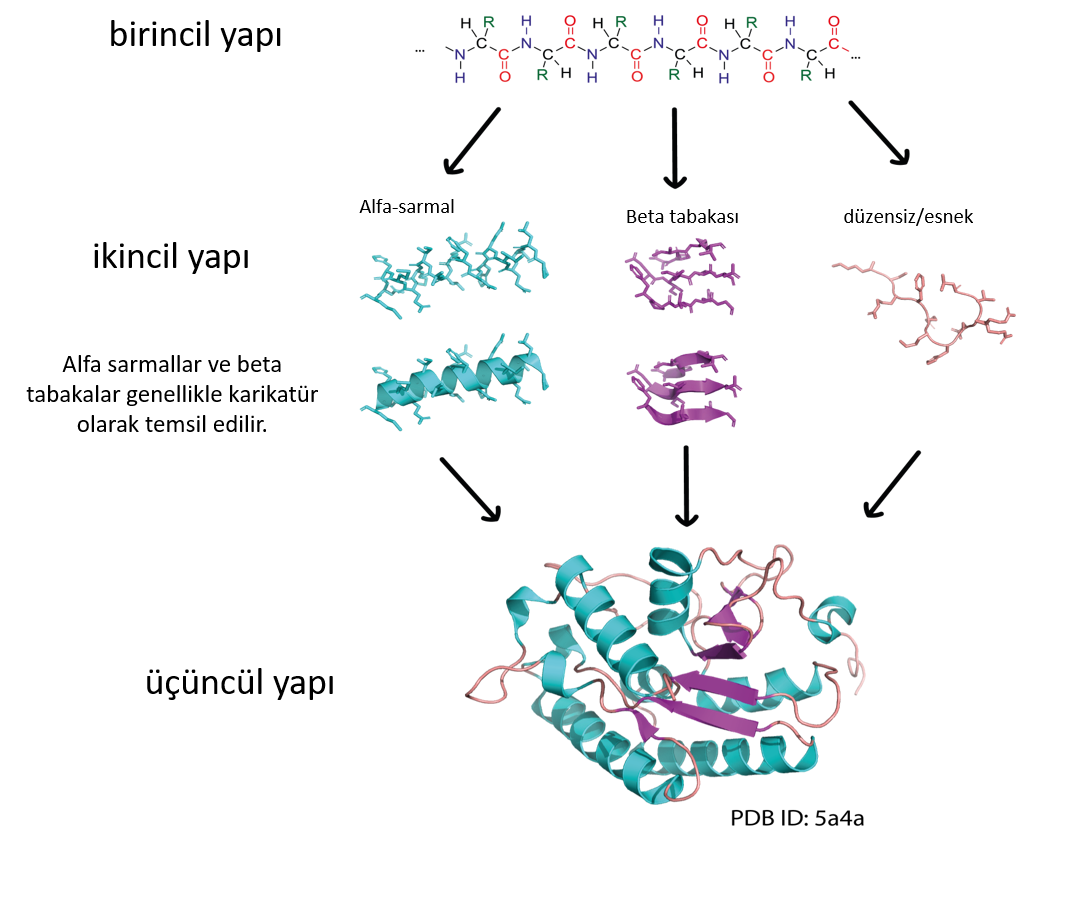
Resim Simone Heber tarafından sağlanmıştır
Bir proteinin 3 boyutlu yapısı işlevini belirler ve katlanma yanlış giderse hataya ve hastalığa yol açabilir. Alzheimer ve Parkinson hastalığı gibi hastalıklar yanlış katlanmış proteinlerle bağlantılıdır.
Proteinlerin 3 boyutlu yapılarını çözmek, yaşamın temel işlevlerini anlamanın anahtarıdır ve hastalıklarla mücadeleye yardımcı olabilir. Örneğin, hastalığa bağlı bir proteinin yapısı, etkili bir ilacın tasarımına rehberlik edebilir.
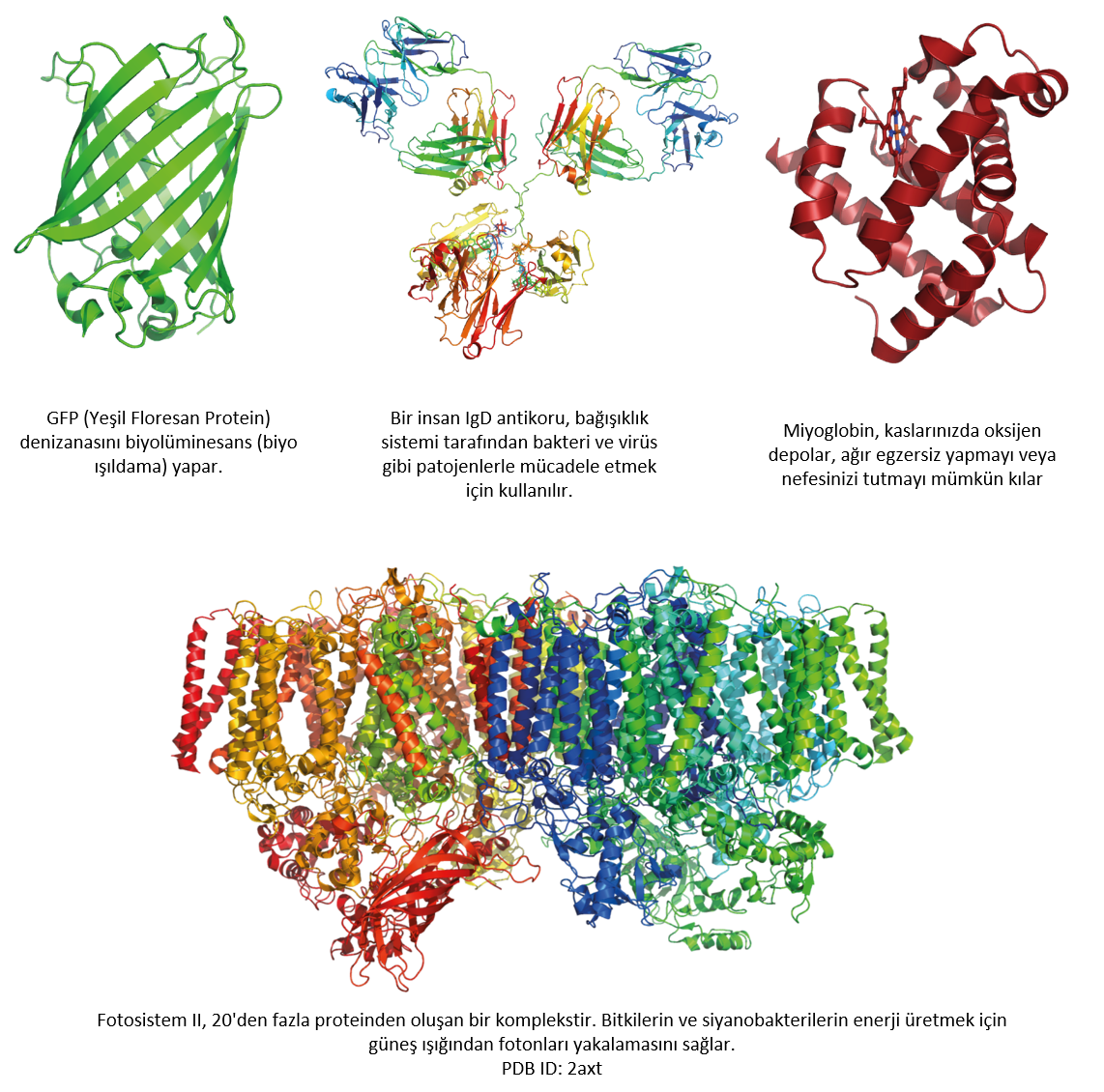
Resim Simone Heber tarafından sağlanmıştır
Protein yapılarını belirlemek için deneysel yöntemler
1962’de Max Perutz ve John Kendrew (daha sonra EMBL’nin kurucularından biri ve ilk direktörü oldu), X-ışını kristalografisi ile ilk kez bir proteinin (miyoglobin) yapısını belirledikleri için Nobel Kimya Ödülü’nü aldılar.[1]
Her ne kadar bazı üniversitelerde daha az güçlü aletler kullanılsa da, X-ışını kristalografisi (örneğin çok parlak X-ışınları yayan daha güçlü senkrotron ışık kaynakları) ile protein kristallerinin çekilmesine dayanır. Protein kristali, X-ışınlarını kırar ve kırınım modeli, bilim adamlarının proteinin yapısını hesaplamasına izin verir.

Christian Hendrich, GNU Özgür Belgeleme Lisansı, Sürüm1.2

PSI SLS, Villigen, CH
Resim Simone Haber’in izniyle
Resim Simone Haber’in izniyle
İkinci bir yöntem, atom çekirdeğinin manyetik alanlarını ölçen nükleer manyetik rezonans (NMR) spektroskopisidir. Bu manyetik alanlar atomların çevresinden etkilenir, bu nedenle proteinlerdeki atomlar için hangi amino asitlerin birbirine yakın olduğu hakkında bilgi verirler. Bu çalışma için 2002 Nobel Kimya Ödülü verildi.[2]

Resim Simone Heber’in izniyle.
Üçüncü bir güçlü yöntem, kriyojenik elektron mikroskobu (kriyo-EM) olup, 2017 Nobel Kimya Ödülü’nün[3] verildiği hızlıca dondurulmuş numuneler üzerinde gerçekleştirilir. Elektron mikroskobunda, ışık yerine bir elektron demeti kullanılır, bu da daha küçük ayrıntıların belirlenmesini sağlar.
Bu yöntemler, bilim adamlarının 150.000’den fazla protein yapısını belirlemesine ve bunları Protein Veri Bankası’nda (PDB)[4] halka açık hale getirmesine izin verdi. Bununla birlikte bu teknikler yavaş, pahalı ve genellikle proteinin doğası ile sınırlıdır. Bilim adamları, herhangi bir başarı garantisi olmadan bir protein yapısını çözmeye çalışmak için yıllarını harcayabilirler.
Neden bir proteinin 3 boyutlu yapısını birincil yapıdan hesaplayamıyoruz?
1972’de Christian B. Anfinsen, bir proteinin birincil dizisinin onun yapısını belirlediğini gösterdiği için Nobel Kimya Ödülü’nü aldı.[5] Bir proteinin 3 boyutlu yapısını amino asit dizisinden tahmin etme fikri yaklaşık 50 yıldır var ve insan DNA dizisinin ortaya çıkarılmasıyla proteinlerin birincil yapıları yaygın olarak mevcut. Peki neden şimdiye kadar çok az başarı elde edildi?
Bir amino asit zinciri teorik olarak çok sayıda üçüncül yapıya katlanabilir: 100 amino asitlik bir protein için, tahmini 10300 (yani arkasonda 299 sıfır olan bir 10!) olası yapı vardır. Doğada, proteinler genellikle en kararlı yapıya katlanır. Bu minimum enerji yapısı hesaplanabilir, ancak tüm olası yapıları karşılaştırmak muazzam miktarda bilgi işlem gücü gerektirir. “Katlanma sorununu” çözme çabaları, gönüllülerden bilgi işlem gücü ödünç alınarak dünyanın en hızlı bilgi işlem sistemlerinden biri haline gelen dağıtılmış bilgi işlem projesi Folding@home’u,[6] içeriyor. Bilgisayarınızdaki, akıllı telefonunuzdaki veya PlayStation3’ünüzdeki kullanılmayan CPU’yu protein katlanmasına katkıda bulunarak dahil olabilir ve bilim adamlarına yardımcı olabilirsiniz!
CASP yarışması ve 2020 kazananı AlphaFold
Protein Yapısı Tahmininin Kritik Değerlendirmesi (CASP) forumu 1994’te kuruldu[7] ve bilim adamlarının deneysel olarak çözülmüş ancak henüz yayınlanmamış protein yapılarını tahmin etmek için yazılım kullandığı iki yılda bir bir yarışma düzenliyor. Ancak şimdiye kadar hiçbiri doğru protein yapısı tahmini sağlayamadı.
2018 yılında DeepMind[8,9] yapay zeka (AI) sistemi AlphaFold ile yarışmaya katıldı ve kazandı. 2020 yarışmasında AlphaFold2, protein yapılarının %90’ından fazlasını deneysel doğruluta tahmin ederek rakiplerini geride bırakarak büyük bir Atılım yaptı.[9–11]
DeepMind, satranç, Go ve StarCraft gibi oyunlarda insanları yenebilen yapay zeka sistemleriyle tanınan Google’ın sahip olduğu bir şirkettir. 2017 yılında AI AlphaGo, dünyanın en iyi Go oyuncusunu yendi. Yapay zeka daha sonra yeniden tasarlandı ve herhangi bir insan katkısı olmadan satrancı öğrendi.
AlphaFold2, derin öğrenmeye dayalı bir yapay zekadır; yüzlerce bilim insanının çalışmalarından yararlanılarak yapay zekaya eğitim için 100.000’den fazla bilinen protein kıvrımı biçimleri yüklendi. Daha sonra, yalnızca birkaç gün içinde doğru protein yapılarını tahmin etmek için eğitim setinden öğrenilen modelleri kullanır.
Bilime ve topluma etkisi nedir?
DeepMind, insan hastalıkları ile ilişkili proteinlerin yapılarını çözmek ve ilaç tasarımına yardımcı olmak için AlphaFold’u kullanmayı planladığını söylüyor. Yapısal bilgi aynı zamanda plastikleri parçalayan veya biyoyakıt üreten enzimlerin mühendisliğine de yardımcı olabilir. Protein yapılarını deneysel olarak çözmek pahalı ve zaman alıcı olduğundan, protein yapısının doğru tahmini, bu tür araştırmaları büyük ölçüde hızlandırabilir ve maliyetini düşürebilir.
Resim Simone Heber’in izniyle
Ancak, makine öğrenimi yöntemleri yapı tahminleri sağlasa da proteinlerin nasıl katlandığını açıklayamadığı için hala açık sorular var. Eğer protein 10300 olası yapının tümünü deneseydi, katlanması evrenin yaşından daha uzun bir süreye ihtiyaç duyardı, ancak doğada proteinler milisaniyeler içinde katlanabiliyor. Bu durum, 1969’da bunu öne süren Cyrus Levinthal’den sonra “Levinthal paradoksu” olarak anılıyor.
Ayrıca, protein yapıları, örneğin başka bir proteine veya bir ilaca bağlanırken belirli bir esnekliğe sahiptir, dolayısıyla bunların katlanması, birincil diziden daha fazlasına bağlıdır. Bu, doğru yapı tahmini olsa bile deneysel yapı tespiti ve fonksiyonel araştırmanın hala önemli olacağı anlamına gelir.
Ancak doğru yapı tahmini, bilimsel ilerlemeyi hızlandırma ve birçok maliyetli, sıkıcı deneyden tasarruf etme potansiyeline sahiptir. Tahminler deneylerin tasarımına rehberlik edebilir, bilimsel süreci kolaylaştırabilir ve bilim adamlarının daha gelişmiş problemleri daha hızlı çözmelerine olanak sağlayabilir.
References
[1] Kimya Nobel dersi 1962, Hızlı okuma: https://www.nobelprize.org/prizes/chemistry/1962/speedread/
[2] Kimya Nobel dersi 2002, Basın açıklamsı: https://www.nobelprize.org/prizes/chemistry/2002/press-release/
[3] Kimya Nobel dersi 2017, Basın açıklamsı: https://www.nobelprize.org/prizes/chemistry/2017/press-release/
[4] Protein veri bankasının ana sayfası: http://www.rcsb.org/
[5] Kimya Nobel dersi 1972, Basın açıklamsı: https://www.nobelprize.org/prizes/chemistry/1972/press-release/
[6] Folding@home bilgisayar projesinin ana sayfası: https://foldingathome.org/
[7] Protein Yapısı Tahmin Merkezi: https://predictioncenter.org/
[8] Alphafold web sayfası: https://deepmind.com/research/case-studies/alphafold
[9] Alphafold üzerine Nature’da bir haber makalesi: https://www.nature.com/articles/d41586-020-03348-4
[10] MIT technology review incelemesi: https://www.technologyreview.com/2020/11/30/1012712/deepmind-protein-folding-ai-solved-biology-science-drugs-disease/
[11] Bir araştırmacının AlphaFold2’ye bakış açısı: https://www.asbmb.org/asbmb-today/science/120520/ai-makes-huge-progress-predicting-how-proteins-fol
Resources
- Çevrimiçi biyolojik veri tabanları ile evrim ve biyokimya öğretimi: Tenorio G (2014) Using biological databases to teach evolution and biochemistry. Science in School 29:30–34.
- Yeşil floresan proteinin yapısının ışık yayma özelliklerini nasıl belirlediğini keşfedin: Furtado S (2009) Painting life green: GFP. Science in School 12:19–23.
- Protein kristalizasyonu ve ESRF hakkında daha fazlası: Cornuéjols D (2009) Biological crystals: at the interface between physics, chemistry and biology. Science in School 11:70–76.
- EMBL-EBI’de biyoinformatik veri depolama hakkında bilgi: Stroe O (2018) Bioinformatics: the new ‘cabinet of curiosities’. Science in School 44:20–24.
- Protein yapısı ve katlanmasıyla ilgili bir karikatür: https://www.youtube.com/watch?v=hok2hyED9go.
Institutions
Author(s)
Dr Simone Heber, Heidelberg’deki EMBL’de doktora sonrası araştırmacı olarak görev yapıyor ve burada oositlerin gelişimi sırasında proteinler ve RNA arasındaki etkileşimleri anlamaya çalışıyor. Doktora derecesini yapısal biyoloji laboratuvarında aldı; burada X-ışını kristalografisini ve NMR’yi kullanarak bir nöron proteininin belirli RNA’ları nasıl tanıyabildiğini ve böylece hafızaya ve öğrenmeye nasıl katkıda bulunduğunu anladı.

Review
Alzheimer, kanser veya bulaşıcı hastalıklar gibi hastalıkların tedavisi büyük ölçüde yeni ilaçların tasarımına yönelik moleküler hedeflerin belirlenmesine bağlıdır. Bilgisayar teknolojileri ve yapay zeka, bunu yapmak için gereken zamanı ve parayı azaltıyor. Bu makale öğrencilerimize hastalıklarla daha etkili bir şekilde mücadele etmenin bir yolu olarak araştırmada disiplinler arası işbirliğinin özünü öğretiyor.
Makale, moleküler 3 boyutlu yapıların, özellikle de proteinlerdeki amino asit dizilerinin katlanmasının önemini ve yapay zeka ile biyoinformatiğin, hücrelerimizdeki ve moleküllerimizdeki gizli bilgileri ortaya çıkarmamıza nasıl yardımcı olabileceğini tanıtmak amacıyla biyoloji ve kimya öğretiminde yararlı olacaktır.
Jesús López Alonso, Biyoloji ve Jeoloji öğretmeni, IES Gil y Carrasco-Ponferrada, İspanya