Supporting materials
Bioinformatyka z kartką papieru i długopisem: jak budować drzewo filogenetyczne (Word)
Download
Download this article as a PDF





Tłumaczenie Jadwiga Schreiber. Analizy bioinformatyczne wymagają zazwyczaj sprawnie działającego komputera. Mimo to Cleopatra Kozlowski pokaże nam jak przeprowadzić taką analizę i odnaleźć naszego ewolucyjnego przodka używając tylko kartki papieru i długopisu.
Dzięki najnowszym osiągnięciom technologicznym stosunkowo łatwo i szybko można ustalić sekwencje DNA czy białka. Same sekwencje jednak niewiele nam mówią, na przykład: GAATCCA. Musimy wiedzieć co one oznaczają. Na przykład: Jakie białko jest kodowane przez daną sekwencję DNA? Czy ona w ogóle koduje jakieś białko? Jaki wpływ miałaby zmiana w sekwencji DNA na strukturę tego białka? Jaką funkcje ma to białko w komórce? Czy ta sekwencja DNA mówi nam coś o naszej ewolucji? A jeśli tak to co?
Odpowiedź na tego typu pytania oraz inne zagadnienia biologiczne można uzyskać za pomocą bioinformatyki: najczęściej poprzez porównanie sekwencji DNA czy białek – na przykład porównując nowo odkryte sekwencje z tymi, o których już stosunkowo dużo wiemy (być może mają one podobną funkcję?) lub porównując podobne sekwencje pomiędzy gatunkami.
Oczywiście bioinformatyka wymaga sprawnie działającego komputera. Jednak pozwalając komputerowi wykonać całą pracę nie zrozumiemy zasad funkcjonowania takich analiz. Dlatego też proponowane tutaj ćwiczenie jest do wykonania na papierze, by uczniowie mogli zrozumieć jak naprawdę działają analizy bioinformatyczne.
Artykuł ten zawiera jedno z czterech ćwiczeń. Dwa wprowadzające ćwiczenia (“Gene finding” i “Mutations”) oraz bardziej zaawansowane ćwiczenie („Mobile DNA”) można pobrać ze strony internetowej European Learning Laboratory for the Life Sciences (ELLS)w1. Wymagane do tych zadań tabele, objaśnienia ćwiczeń krok po kroku oraz propozycje pytań sprawdzających zrozumienie tematu można pobrać ze strony internetowej Science in Schoolw2.
Akumulacja mutacji powoduje zmianę sekwencji DNA poprzez pokolenia. Przedstawione tutaj ćwiczenie zademonstruje nam jak możemy to wykorzystać by ustalić ewolucyjną zależność (odległość) między różnymi organizmami. Zajmie to ok 90 minut i wymaga niczego więcej jak tylko wspomnianych wyżej tabeli i długopisuw2.
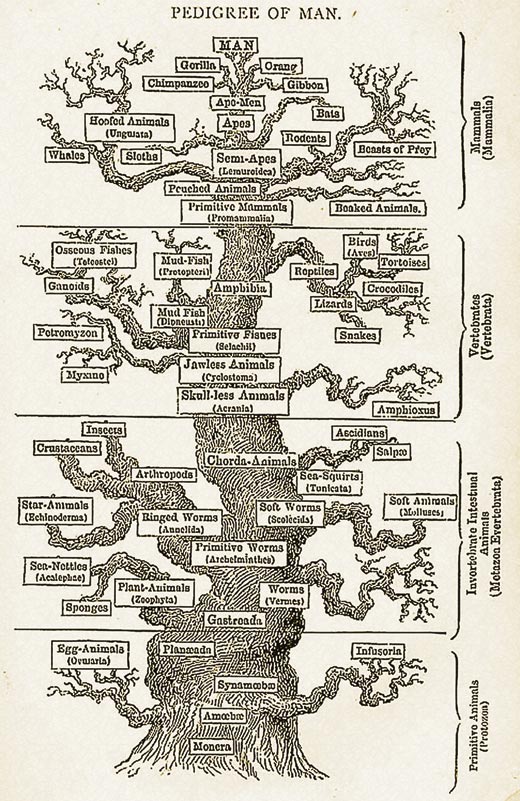
Jak sklasifikowałbyś różnego rodzaju zwierzęta? Tradycyjnie, ewolucyjną zależność pomiędzy zwierzętami określano na podstawie ich różnic morfologicznych na przykład posiadanie kręgosłupa czy skrzydeł. Taki podział może być jednak problematyczny. Ptaki, nietoperze i owady mają skrzydła ale czy to sprawia, że są one blisko spokrewnione? Jak można zatem dowiedzieć się w jakim stopniu różne organizmy są ze sobą spokrewnione i kiedy oddzieliły się one od wspólnego przodka?
Z badań DNA wiemy, że mutacje w materiale genetycznym powstają przypadkowo i stosunkowo rzadko oraz że są one przekazywane do następnego pokolenia. Zatem jeśli założymy, że wszyscy pochodzimy od wspólnego przodka, to dzięki porównaniu sekwencji homologicznych możemy oszacować kiedy poszczególne organizmy oddzieliły się od niego. Lub innymi słowami, im więcej czasu upłynęło od momentu oddzielenia się dwóch gatunków od wspólnego przodka tym więcej różnic znajdziemy w ich DNA.
Sekwencjami homologicznymi nazywamy te sekwencje DNA u dwóch różnych gatunków, które mają wspólne pochodzenie. W rzeczywistości nie mamy żadnego dowodu, że w ogóle jakaś sekwencja jest homologiczna – nie byliśmy świadkami zmian zachodzących w DNA. Jednak jeśli dwie sekwencje są wystarczająco do siebie podobne, zakładamy, że są one „homologiczne”. Żeby sprawdzić w jakim stopniu dwie sekwencje są do siebie podobne, trzeba je odpowiednio przyporządkować i porównać.
Zauważ, że różne regiony DNA – kodujące i niekodujące – ewoluowały w różnym tempie. Kodujące regiony ewolują (zmieniają się) stosunkowo wolno. Dlaczego? Jeśli mutacja spowoduje zmianę w białku (np. strukturze białka) to może to być dość kosztowne dla organizmu – być może zmniejszy to szansę jego przeżycia i/lub pozostawienie po sobie potomstwa. Temat ten omówiony jest w zadaniu „Mobile DNA.
By zailustrować zasadę homologii, skorzystamy z filologii (lingwistiki) – nauki o ewolucji języków. Istnieje bowiem wiele podobieństw w metodach używanych do badań ewolucji języków i ewolucji organizmów.
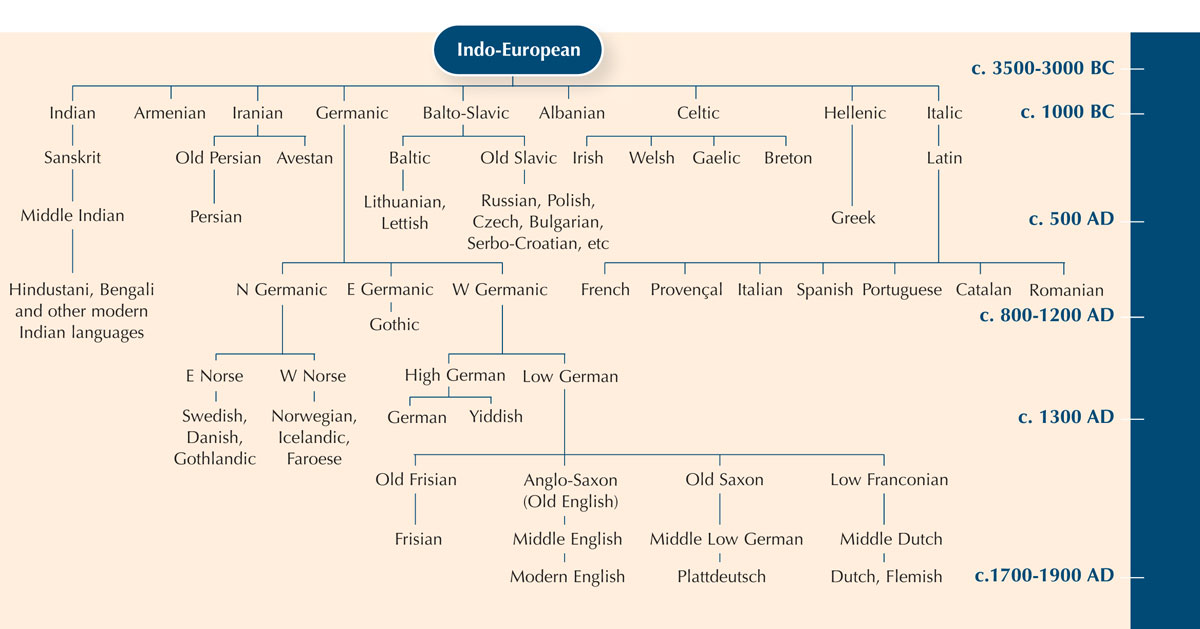
Szukanie różnic we fragmentach DNA pomiędzy organizmami jest jak porównywanie słowa znaczącego to samo w różnych językach, by zbadać jak blisko języki te są ze sobą spokrewnione.
| Ormiański | gatz |
| Baskijski | katu |
| Niderlandzki | kat |
| Angielski | cat |
| Estoński | kass |
| Fiński | kissa |
| Islandzki | kottur |
| Włoski | gatto |
| Norweski | katt |
| Polski | kot |
| Portugalski | gato |
| Rosyjski | kot |
| Hiszpański | gato |
| Szwedzki | katt |
Na pewno zwróciłeś uwagę, że słowo “kot” po włosku, hiszpańsku i portugalsku brzmi prawie tak samo: gatto, gato i gato. W języku szwedzkim i norweskim kot jest „katt” ale zwróć uwagę, że w języku fińskim to „kissa”. Choć Finlandia jak Szwecja i Norwegia jest krajem nordyckim, fińskie słowo kot jest bardziej podobne to estońskiego słowa „kass”. Rzeczywiście języki te są ze sobą spokrewnione. Zatem, porównując jak konkretne słowo zmieniło się z czasem i regionem, możemy oszacować związek pomiędzy różnymi językami.

W zadaniu tym skonstrułujemy drzewo filogenetyczne używając pięciu homologicznych sekwencji DNA należących do ssaków naczelnych. Ponieważ sekwencje te są fikcyjne, nie możemy użyć ich do określenia prawdziwego dystansu genetycznego tych ssaków; aby zbudować prawdziwe drzewo filogenetyczne wymagane są dużo dłuższe sekwencje genetyczne. Nie mniej jednak te fikcyjne sekwencje (patrz tabelka nr. 2) zostały tak dobrane by ukazać nam w miarę rzeczywiste powiązanie pomiędzy tymi gatunkami.
Uwaga: wszystkie potrzebne do wykonania tego zadania tabelki można pobrać ze strony internetowej Science in Schoolw2.
| Ssaki naczelne | Sekwencje |
|---|---|
| Neandertalczyk (n) | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
| Człowiek (h) | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Szympans (c) | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
| Goryl (g) | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
| Orangutan (o) | ACAACCTGCACTCCTATTCTGCCGAGCCGGGCGCGTGGCAAAGTCC |
| Neandertalczyk | TGGTCCTGCAGTCCTCTCCTGGCGCCCCGGGCGCGAGCGGTTGTCC |
|---|---|
| Człowiek | TGGTCCTGCTGTCCTCTCCTGGCGCCCTGGGCGCGAGCGGATGTCC |
| Szympans | TGATCCTGCAGTCCTCTTCTGGCGCCCTGGGCGCGTGCGGTTGTCC |
|---|---|
| Goryl | TGGACCTGCAGTCATCTTCTGCCCGCCCGAGCGCTTGCCGATGTCC |
Tabele porównawcze dla wszystkich par gatunków oraz tabelę sumującą różnice między nimi można pobrać ze strony internetowej Science in Schoolw2.
| Neandertalczyk | Człowiek | Szympans | Goryla | Orangutan | |
|---|---|---|---|---|---|
| Neandertalczyk | 0 | 3 | |||
| Człowiek | 3 | 0 | |||
| Szympans | 0 | 11 | |||
| Goryl | 11 | 0 | |||
| Orangutan |
Dzieląc liczbę różnic w nukleotydach w sekwencjach pomiędzy dwoma gatunkami przez całkowitą liczbę nukleotydów w każdej z sekwencji (w tym wypadku 46) uzyskamy stosunkową odległość genetyczną między tymi sekwencjami (gatunkami).
Zakłada się, że “średnia różnica” w sekwencjach dwóch gatunków przedstawia ich przodka. W tym ćwiczeniu nie obliczamy bezpośrednio średniej różnicy pomiędzy np. Neandertalczykiem i człowiekiem, a ewolucyjną odległość pomiędzy przodkiem Neandertalczyka / człowieka i wszystkimi innymi naczelnych z naszej grupy.
| Neandertalczyk/ człowiek | Szympans | Goryl | Orangutan | |
|---|---|---|---|---|
| Neandertalczyk/ człowiek | 0 | (4+5)/2 = 4.5 | (11+12)/2=11.5 | |
| Szympans | (4+5)/2 = 4.5 | 0 | ||
| Goryl | (11+12)/2=11.5 | 0 | ||
| Orangutan |
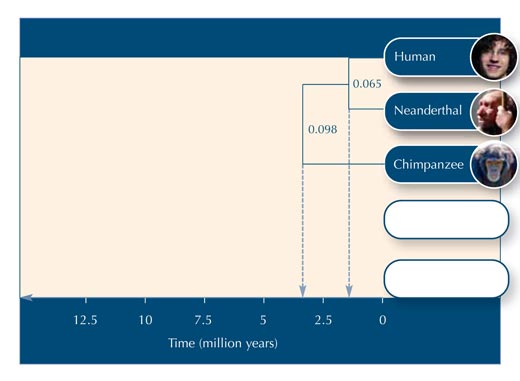
Teraz na podstawie danych z tabeli nr. 5, możemy budować drzewo ewolucyjne (zobacz rysunek nr. 2).
Załużmy, że 20 milionów lat musi upłynąć dla jednego nukleotydu, w danej sekwencji DNA, by uległ on zmianie. Aby sekwencja DNA zmieniła się o 0.065, upłynie 0.065*20 milionów = 1.3 milionów lat. Zatem długość tej gałęzi odzwierciedla 1.3 milionów lat na skali czasu (zobacz rysunek 2).
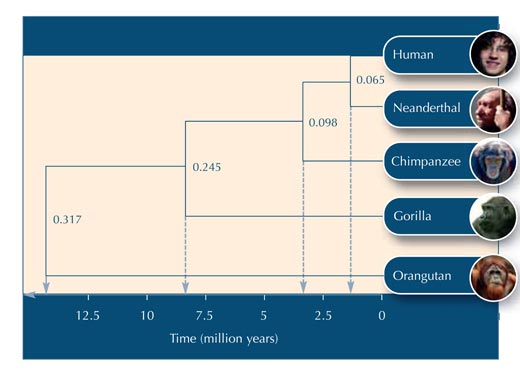
BPoniżej znajdują się przykładowe pytania sprawdzające zrozumienie przez uczniów powyższego ćwiczenia. Proponowane odpowiedzi są do pobrania ze strony internetowej Science in Schoolw2.
Ćwiczenie to zostało opracowane dzięki współpracy European Learning Laboratory for the Life Sciences (ELLS)w1 i European Molecular Biology Laboratory’s E-STAR Fellows – organizacji opracowujących materiały dydatkyczne. Działalność Cleopatry Kozlowski sponsorowana jest przez European Commission’s Framework Programme 6 Marie Curie Host Fellowship for Early Stage Research Training, numer projektu MEST-CT-2004-504640.
Hodge R (2006) A new tree of life. Science in School 2: 17-19.
Kiedy słyszymy o bioinformatyce od razu wyobrażamy sobie skomplikowane programy komputerowe i maszyny sekwencjujące DNA. Ale metody używane w tej stosunkowo nowej dziedzinie naukowej można zaprezentować za pomocą prostych ćwiczeń wymagających tylko kartki papieru i długopisu.
Cleopatra Kozlowski zachęca nas i prezentuje jak zbudować drzewo filogenetyczne człowieka i innych ssaków naczelnych na podstawie różnic genetycznych w krótkich (przykładowych) sekwencjach DNA. Zaproponowane ćwiczenie (zabawa) można użyć jako materiał dydaktyczny do omówienia trudnych tematów biologicznych jak ewolucja czy znaczenie zegara molekularnego.
Artykuł ten zawiera również interesujące propozycje pytań sprawdzających zrozumienie tematu dla nauczycieli i uczniów. Podane referencje internetowe oferują dodatkowe informacje o ewolucji i tematach z nią związanych.
Giulia Realdon, Włochy
Bioinformatyka z kartką papieru i długopisem: jak budować drzewo filogenetyczne (Word)
Download this article as a PDF