




Tödliche Proteine: Prionen Understand article
Übersetzt von Hildegard Kienzle-Pfeilsticker. Seit der Rinderwahn-Epidemie in den 1980er und 1990er Jahren und dem Auftreten ihres menschlichen Pendants, der Creutzfeld-Jacob-Variante der Erkrankung wurde viel über Prionen, die auslösenden Agenzien, geforscht. Mico Tatalovic berichtet über…

/ iStockphoto
Möglicherweise existiert eine versteckte Epidemie mit Millionen von bereits infizierten Menschen, die jederzeit ausbrechen kann; wir können sie nicht verhindern oder die Krankheit heilen und wir können sie erst diagnostizieren wenn die fatalen Symptome auftreten.
Die Krankheit ist eine Variante der Creutzfeld-Jacob-Krankheit (vCJK), einer Erkrankung aus einer Gruppe von Krankheiten, die als transmissible (übertragbare) spongiforme Enzephalopathien (TSE) bekannt sind. Sie werden verursacht und übertragen durch abnorme Formen von Prion-Proteinen. Beispiele für TSE sind nicht nur vCJD, sondern auch Scrapie der Schafe, bovine spongiforme Enzephalopathie (BSE oder Rinderwahn) bei Rindern, Kuru beim Menschenw1 Diese Krankheiten erzeugen große, flüssigkeitsgefüllte Räume im Hirngewebe, weil die Akkumulation fehlgeformter Prion-Proteine das Absterben von Neuronen (Nervenzellen) verursacht. Diese charakteristischen, durchlöcherten Strukturen geben den Krankheiten ihre Namen: spongiforme (wie ein Schwamm) Enzephalopathien (Hirnkrankheiten).
TSEs beeinträchtigen das zentrale Nervensystem, wodurch Symptome wie Koordinations- und Balanceprobleme, Unsicherheit und ruckartige Bewegungen auftreten. Beim Menschen verursachen TSEs auch Persönlichkeitsveränderungen und Depressionen, Betroffene können unter Verwirrung, Gedächtnisproblemen und Schlaflosigkeit leiden. Das Fortschreiten der Krankheit führt zum Verlust der meisten geistigen Fähigkeiten, inklusive der Sprechfähigkeit. Alle TSEs führen zum Tod und eine Therapie ist noch nicht in Sicht.

proteins (orange) from an animal
infected with scrapie
Image courtesy of R. Dourmashkin
/ Wellcome Images
Prionen sind spezifische Proteine, die man vor allem im Nervensystem findet, wo sie in ihrer normalen Form wichtige Funktionen übernehmen können. Beispielsweise legen Studien an der Meeresschnecke Aplysia nahe, dass Prionen eine entscheidende Rolle bei der Ausbildung des Gedächtnisses haben (Si et al., 2010). Infektiöse Prionen sind abnormale (aberrante, fehlgeformte) Formen von Prion-Proteinen, die sich innerhalb ihres Wirtes vervielfältigen, indem sie Proteinen desselben Typs ihre fehlgeformte Struktur aufzwingen. Dadurch wird ein Dominoeffekt ausgelöst, durch den eine kleine Zahl fehlgeformter Prionen viele normale beeinträchtigen kann und schließlich zur Krankheit führt. Wenn die fehlgeformten Prionen in der Zelle Amyloide – Proteinaggregate – bilden, sterben die Zellen ab und hinterlassen durchlöcherte Strukturen im Gehirn.
Prionen sind die einzigen bekannten, sich selbst verbreitenden pathogenen (krankheitsauslösenden) Proteine und sie sind in der Lage eine schwere Krankheit auszulösen, obwohl sie scheinbar nur Proteinmoleküle sind: anders als bei Bakterien, Viren oder anderen bekannten Pathogenen ist in ihnen keine Information in Nukleinsäuren (wie DNA oder RNA) gespeichert, die sagt, wie sie in ihrem Wirt eindringen und sich darin vervielfältigen sollen. Prionen umgibt immer noch ein Geheimnis und es ist unklar, wie genau sie sich vervielfältigen, wie sie die Blut-Hirn-Schranke und die Gattungsbarriere überwinden – das heißt, wie sie verschiedene Wirtsgattungen infizieren können.

Robert Koch (1843-1910)
In den 1960er Jahren merkten Forscher zum ersten Mal, dass TSE auslösende Agenzien scheinbar keine Nukleinsäuren besaßen; Tikvah Alper schlug vor, dass das Agens ein Protein sei. Diese Idee klang herausfordernd, weil alle anderen bekannten krankheitsauslösenden Agenzien Nukleinsäuren enthielten und ihre Virulenz und Pathogenese genetisch bestimmt waren. Nach weiteren drei Jahrzehnten Forschungsarbeit, verfolgt vor allem durch Stanley Prusiner, der 1997 den Nobelpreis für Physiologie oder Medizin für seine Arbeiten mit Prionen und TSEs erhielt, wurde die „Nur-Protein-Hypothese“ weithin anerkanntw2.
Trotzdem gibt es immer noch Leute, die glauben, dass Prionen-Krankheiten in Wirklichkeit durch unkonventionelle Viren verursacht werden und dass Prionen-Proteine nur Teil dieses mysteriösen Virus sind. Die Kochschen Postulate beschreiben die zu führenden Nachweise für ein bestimmtes Agens, um zu zeigen, dass es eine bestimmte Krankheit auslöst; ein notwendiger Schritt ist das Auslösen der Krankheit durch das Agens in einem gesunden Organismus. Um zu beweisen, dass TSE tatsächlich durch das Prion-Protein selbst verursacht wird, müssen isolierte, gereingte, fehlgeformte Prionen zur Übertragung der Krankheit eingesetzt werden. Genau so wurde das erst im Februar 2010 durchgeführt und weitere wichtige Beweise zur Nur-Protein-Hypothese ergänzt (Wang et al., 2010).
Warum haben manche Wissenschaftler Angst vor einer vCJD-Epidemie?
Der Prionen-Stamm, der am meisten Sorgen bereitet, ist der, der vCJD auslöst – eine Form des Rinderwahns, die die Gattungsgrenzen überschritten und Menschen infiziert hat (siehe Kasten).der erste Fall von vCJD tauchte im UK 1996 auf und hat dort seither 168 Menschen getötet, mit dem Gipfel im Jahr 2000, als 28 Menschen starbenw3, Weniger Menschen starben daran in anderen Ländern wie Frankreich, Italien, Irland, Kanada und den USA. Die genauen Zahlen Infizierter oder Verstorbener aufgrund dieser Krankheit weltweit sind nicht bekannt und obwohl immer och Unsicherheit darüber herrscht, wie man sich ansteckt, geht man davon aus, dass der Hauptinfektionsweg der Genuss infizierten Rindfleisches ist.
| UK | 172 (4) |
|---|---|
| Frankreich | 25 (0) |
| Republik Irland | 4 (0) |
| Italien | 2 (1) |
| USA |
3 (0) |
| Kanada | 1 (0) |
| Saudi Arabien | 1 (1) |
| Japan | 1 (0) |
| Niederlande | 3 (0) |
| Portugal | 2 (0) |
| Spanien | 5 (0) |
Die meisten anderen Prionen-Krankheiten, „normale“ CJD eingeschlossen, haben genetische Ursachen oder treten ohne erkennbare Gründe auf und betreffen ältere Leute. Die vCJD unterschiedet sich von CJD, weil sie junge Leute betrifft und man die fehlgeformten Prionen nicht nur im Gehirn, sondern auch in Geweben wie Blut, Zunge und Blinddarm findet; neue Übertragungswege in der gesamten Population werden dadurch möglich, nicht nur bei älteren Leuten.
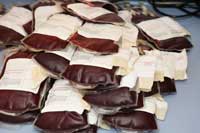
Beispielweise könnten Prionen durch Bluttransfusionen übertragen werden. Im UK wird gespendetes Blut an Leukozyten verarmt (möglicherweise mit infektiösen Prionen infizierte Leukozyten werden entfernt). Viele andere Länder (z.B. Deutschland) verbieten Blutspenden von Menschen, die im UK gelebt haben.
Die Krankheit könnte auch durch chirurgische Instrumente verbreitet werden: infektiöse Prionen sind resistent gegen hohe Temperaturen, Strahlung und übliche chemische Behandlungen, die andere bekannte Pathogene zerstören. Die Schätzungen über die Höhe des Risikos gehen auseinander.
Weiter fand man Prionen auch in der Epididymis und Samenflüssigkeit von Schafböcken (Gatti et al., 2002). Solche Befunde ließen Bedenken bezüglich Prionen-Infektionen durch Spermaspenden aufkommen und führten sogar zum Verbot von Spermaspenden durch Europäer und Männer, die in Europa gelebt hatten. Ein weltweites Expertengutachten schätzte die Chance einer Prionen-Übertragung über Sperma auf weniger als 1:10 000 000 ein (Mortimer & Barratt, 2006).
Aber Gewebekontakt – sei es über Sperma oder Blut oder über chirurgische Instrumente – ist nicht die einzig mögliche Übertragungsquelle: die meisten von uns konsumieren Milch und Milchprodukte.
Mit freundlicher Genehmigung
von choja / iStockphoto
Mit freundlicher Genehmigung
von UCL / Wellcome Images
Mit freundlicher Genehmigung
von ImageMediaGroup / iStockphoto
In 2006, a team of scientists from Switzerland detected low levels of the normal form of prions in milk bought in European shops (Franscini et al., 2006). Another study found that aberrant prions were able to replicate within mammary glands of scrapie-infected sheep (Ligios et al., 2005). Taken together, these results suggest that aberrant prions might be present in the milk of animals suffering from prion diseases. As prion disease symptoms take several years to develop, aberrant prions may be present in milk that is sold before the animals are diagnosed with the illness. Nonetheless, although it is not yet possible to give a clear estimate of the risk, it is generally thought that milk is safe until proven otherwise.

eine Infektion mit Prionen:
Milch
Mit freundlicher Genehmigung
von DaveAlan / iStockphoto
It seems, therefore, that we could contract vCJD via several routes although the risk is probably low. But are we all equally at risk? Several studies have shown that vCJD only affects people with certain variants (alleles) of a particular gene (prnp); other people seem to be resistant or do not develop the symptoms as fastw4. Worryingly, about 40% of the Western European and North American population and up to 92% of the Japanese population are homozygous for these susceptibility alleles. Furthermore, research on kuru suggests that even individuals without the susceptibility allele may become infected but simply take much longer to develop symptoms. So perhaps we are all susceptible to prion infection.
Because of all of these possible transmission routes and uncertainties in how important they are, the results of studies predicting the number of people dying from vCJD in the UK range from several hundred to more than ten million people. Even though it now looks as though the vCJD epidemic in the UK is diminishing, this may be just because many infected people are still developing symptoms. No one knows for sure. Ongoing research will elucidate just how risky prion diseases are to public health.
BSE (mad cow disease)
BSE came to scientists’ attention in 1986 when the first cases of a new neurological disease in cattle were discovered in the UK. The cause was traced to the use of beef products in cattle feed. In the resulting BSE epidemic, 181 376 cases of BSE were recorded in the UK by November 2002 and millions of cattle were culled to halt the epidemic. The first cases outside the UK appeared in 1989 and since then there have been several thousand cases in other countries, including most of the European countries, the USA, Canada, Japan and Israel. However, cattle feed with bovine parts was banned in the UK in 1988 and there are strict monitoring procedures in place to keep track of BSE.
Cases of BSE peaked in the UK in 1992 (37 280 cases) and have decreased dramatically since then (12 cases in 2009). The number of cases in other countries peaked some years later (2001-03) but has also decreased dramatically since thenw5.
References
- Franscini N et al. (2006) Prion protein in milk. PLoS ONE 1: e71. doi: 10.1371/journal.pone.0000071
- All PLos ONE articles are freely available online.
- Gatti JL et al. (2002) Prion protein is secreted in soluble forms in the epididymal fluid and proteolytically processed and transported in seminal plasma. Biology of Reproduction 67: 393-400. doi: 10.1095/biolreprod67.2.393
- Ligios C et al. (2005) PrPSc in mammary glands of sheep affected by scrapie and mastitis Nature Medicine 11: 1137–1138. doi: 10.1038/nm1105-1137
- Mortimer D, Barratt CLR (2006) Is there a real risk of transmitting variant Creutzfeldt–Jakob disease by donor sperm insemination? Reproductive BioMedicine Online 13: 778–790
- To view articles in Reproductive BioMedicine Online, it is necessary to register, but registration is free. See: http://www.rbmojournal.com/
- Si K et al. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140: 421-435. doi: 10.1016/j.cell.2010.01.008
- Wang F et al. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327: 1132-1135. doi: 10.1126/science.1183748
Web References
- w1 – Informationen über Kuru erhält man auf der Webseite des US National Institute of Neurological Disorders and Stroke: www.ninds.nih.gov/disorders/kuru
- 1976 erhielt Carleton Gajdusek den Nobelpreis für Physiologie oder Medizin für seine Arbeiten über Kuru. Mehr Informationen sind zugänglich in der Presseerklärung, seiner Autobiografie, Nobelpreis-Rede und anderen Materialien auf der Nobelpreis-Webseite. Siehe: http://nobelprize.org/nobel_prizes/medicine/laureates/1976
- w2 – Sie erfahren mehr über die Arbeit von Stanley Prusiner aus der Presseerklärung, seiner Autobiografie und anderen Materialien auf der Nobelpreis-Webseite. Siehe: http://nobelprize.org/nobel_prizes/medicine/laureates/1997
- w3 – Statistische Daten über die berichteten Fälle von BSE und Todesfälle durch vCJD sind auf der Webseite des European Centre for Disease Prevention and Control erhältlich: (www.ecdc.europa.eu) oder via den direkten Link: http://tinyurl.com/yjbx8tn
- w4 – Mehr über die genetische Disposition, Prionen-Krnakheiten zu entwickeln erfährt man auf der Webseite der Prionen-Abteilung des UK Medical Research Council (www.prion.ucl.ac.uk) oder man benutzt den direkten Link: http://tinyurl.com/yaqau4a
- w5 – Statistics on BSE and many other animal diseases are available on the website of the World Organisation for Animal Health: www.oie.int
Resources
- Mehr Informationen über Prionen sind erhältlich auf den Webseiten der US Centers for Disease Control and Prevention: www.cdc.gov/ncidod/dvrd/prions
- Um mehr über BSE zu lernen siehe die Webseite der Weltgesundheitsorganisation: www.who.int/zoonoses/diseases/bse/en
- Möglicherweise interessante wissenschaftliche Publikationen sind:
- Aguzzi A, O’Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery 9: 237-248. doi: 10.1038/nrd3050
- Collinge, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318: 930-936. doi: 10.1126/science.1138718
- Di Guardo G, Marruchella G (2010) Prions and neuronal death. Cell Death and Disease 1: e6. doi: 10.1038/cddis.2009.9
- Welberg L (2010) Prions: a protective role for prions. Nature Reviews Neuroscience 11: 151. doi: 10.1038/nrn2812
- Die Kochschen Postulate wurden im späten 19. Jahrhundert von Robert Koch publiziert. Zu einer neueren Diskussion über die Postulate und ihrer Gültigkeit siehe:
- Evans AS (1976) Causation and disease: the Henle-Koch postulates revisited. The Yale Journal of Biology and Medicine 49: 175-195. The article can be downloaded free of charge from PubMed Central (www.ncbi.nlm.nih.gov/pmc) or via the direct link: http://tinyurl.com/y9t2pd8
Review
Viele Leute haben von vCJD oder Rinderwahn gehört, wissen aber wenig über das auslösende Agens und die Krankheit selbst. Dieser Beitrag erklärt die Krankheit und zitiert aktuelle Forschung über das auslösende Agens, ein Prion. Dies wird für das Thema Infektionskrankheiten vieler wissenschaftlicher Lehrpläne und für das Allgemeinwissen in anderen Themengebieten nützlich sein. Die Links zu BSE-Daten könnten im Mathematikunterricht oder zum Erlernen der Erstellung aussagekräftiger Schaubilder aus Daten genutzt werden. Der artikel eignet sich für Diskussionen über Lebensmittelsicherheit, die Wirkung des Ausbruchs der Krankkheit auf britische Rinderfarmer sowohl als auch Grundlage zum Verständnis und Erweiterung des Wissens. Einige Vorschläge zur Verbesserung des Verständnisses sind:
- Seit wie vielen Jahren wird an Prionen geforscht
-
10 Jahre
-
25 Jahre
-
30 Jahre
-
40 Jahre
-
- Wer gewann den Nobelpreis für seine Forschungen an Prionen?
- Was ist ein Prion?
- Beschreiben Sie die Auswirkungen der spongiformen Enzephalopathien.
- In welchen Geweben wurden Prionen entdeckt?
- Wie könnten sich Prionen verbreiten?
Ideen für weiterführendes Arbeiten sind:
- Forschen Sie dem BSE-Ausbruch im UK in den späten 1980er Jahren nach und beschreiben Sie die ergriffenen Maßnahmen, um ihn zu begrenzen.
- Nennen Sie die Kochschen Postulate. Erklären Sie warum Prionen bis vor kurzem nicht in seine Hypothese gepasst haben.
Shelley Goodman, UK